
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Experimenteller Workflow für eccDNA-Sequenzierung: Anreicherung, Bibliotheksvorbereitung und häufige Fallstricke

Extrachromosomale zirkuläre DNA (eccDNA) ist eine "Nadel im Heuhaufen": seltene zirkuläre Moleküle, die sich vor einem überwältigenden Hintergrund von linearer genomischer DNA verteilen. Ihre zuverlässige Sequenzierung erfordert einen disziplinierten, durchgängigen Workflow und explizite Qualitätskontrollen. In diesem praktischen Leitfaden für Wissenschaftler im Labor werden wir einen vierstufigen eccDNA-Sequenzierungsworkflow durchgehen – Extraktion von hochmolekularer (HMW) DNA, Eliminierung linearer DNA (eccDNA-Anreicherung), Rolling Circle Amplification (RCA) und Bibliotheksvorbereitung – sowie die Kontrollen, Schwellenwerte und Langlesevalidierung, die Ergebnisse in reproduzierbare Befunde umwandeln.
Wenn Sie neu in diesem Thema sind, beginnen Sie mit dem Konzeptüberblick in eccDNA-Sequenzierung erklärt.
Schritt 1: Extraktion von hochmolekularer DNA (HMW)
Standard-Minipreps sind oft unzureichend für die Entdeckung von eccDNA, da sie DNA scheren und die Rückgewinnung auf kleinere Fragmente ausrichten. Große Zirkeln können fragil sein; harsches Lyseverfahren, wiederholtes Vortexen und aggressive Säulenbindung/-elution können sie anritzen oder brechen. Das Ziel hier ist saubere, HMW-DNA mit minimalem Scheren.
Worauf man abzielen sollte
- Intakte HMW-DNA (breiter hochmolekularer Schimmer auf einem Niederspannungs-0,8%-Agarosegel) und genaue dsDNA-Quantifizierung mit Qubit. Vermeiden Sie es, sich ausschließlich auf NanoDrop zu verlassen – Protein-, Phenol- oder Salzübertragungen können das A260/280-Verhältnis verzerren.
- Sanfte Lyse und minimales Pipettieren: denken Sie langsam, kalt und verwenden Sie, wenn möglich, breite Spitzen.
- Festphasen- oder perlenbasierte Aufreinigung, die lange Moleküle bewahrt. Mehrere eccDNA-Studien berichten von robuster HMW-Erhaltung mit perlenbasierten Ansätzen und bestätigen die Integrität über Gel-/TapeStation-Profile.
Vorgeschlagene Eingaben und Handhabung
- Empfohlene Eingabemasse: 0,5–2 µg HMW-DNA pro Probe für die nachgelagerte Verdauung und RCA, abhängig von der Gewebe-/Zellausbeute.
- EDTA in Lysepuffern verwenden, um Nukleasen zu hemmen; längere Inkubationen bei Raumtemperatur vermeiden.
- Lagern Sie intermediäre DNA bei 4 °C für kurze Zeiträume; frieren Sie sie bei −20 °C für längere Lagerung ein. Auftauen auf Eis.
QC-Prüfpunkt 1: Integrität der Extraktion
- Führen Sie 0,8% Agarose bei niedriger Spannung aus; hochmolekulare DNA sollte als breiter oberer Schimmer mit wenigen diskreten niedermolekularen Bändern erscheinen.
- Qubit dsDNA-Konzentration: Dokumentieren Sie Ausbeute und Reinheit. Optional kann die TapeStation/Bioanalyzer die Fragmentverteilung für Ihre Unterlagen erfassen.
Entscheidungsregel
- Wenn das Gel starke Fragmentierung zeigt oder DNA knapp ist (<100 ng), ziehen Sie in Betracht, mit sanfterer Lyse und bead-basierter Reinigung erneut zu extrahieren. Fahren Sie nicht mit der Exonuklease-Digestion fort, wenn die DNA eindeutig degradiert ist; Sie erhöhen die falsch positiven Ergebnisse und verringern die Wiedergewinnung von Zirkeln.
Beweiszusammenhang
- Viele Labore bestätigen die HMW-Integrität mit Gelen/TapeStation vor der Kreis-Sequenzierung. Übersichten fassen bead-basierte Methoden und Integritätsprüfungen für eccDNA-Workflows zusammen, z.B. in der Frontiers in Genetics Methodenübersicht (2024), die Auswahl der Extraktionsmethoden und die nachgelagerte Anreicherung behandelt.
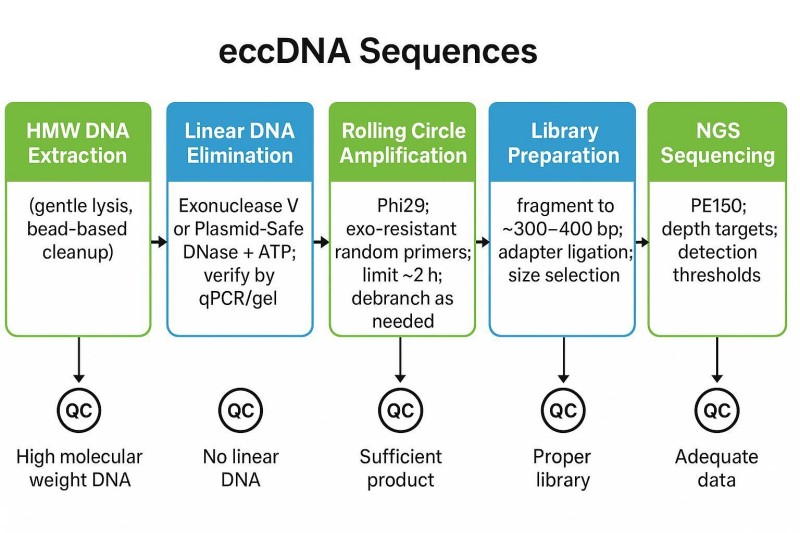 Abbildung 1. eccDNA-Workflow: HMW-Extraktion → Exo-Digestion → RCA → Bibliotheksvorbereitung → NGS. QC-Prüfungen: Extraktionsintegrität, Verdauungseffizienz, RCA-Profil, Bibliotheksmetriken.
Abbildung 1. eccDNA-Workflow: HMW-Extraktion → Exo-Digestion → RCA → Bibliotheksvorbereitung → NGS. QC-Prüfungen: Extraktionsintegrität, Verdauungseffizienz, RCA-Profil, Bibliotheksmetriken.
Schritt 2: Eliminierung linearer DNA (Anreicherung von eccDNA)
Der Grundstein eines eccDNA-Sequenzierung Der Workflow umfasst die rigorose Entfernung von linearer DNA. Zwei Enzymfamilien dominieren diesen Schritt:
- Exonuklease V (RecBCD; häufig von NEB): verdaut lineare dsDNA; die Aktivität hängt von ATP und Puffer ab. Sie kann genickte Ringe abbauen.
- Plasmid-Safe ATP-abhängige DNase (Epicentre/Lucigen): zerstört selektiv lineare dsDNA, während intakte geschlossene zirkuläre dsDNA geschont wird; Circle-Seq-Protokolle bevorzugen oft mehrstündige bis mehrtägige Verdauungen mit ATP-Nachdosierung.
Setups, die in der Praxis funktionieren
- Exonuklease V: bei 1X NEBuffer 4 mit ~1 mM ATP bei 37°C laufen; die Inkubationszeit hängt von der Eingabemasse und dem Hintergrund ab. Die NEB-Dokumentation weist auf die ATP-Anforderung und die selektive Entfernung von linearer DNA hin; siehe NEB Exonuclease V Produktseite und Pufferaktivitätsanleitung.
- Plasmid-Safe: bei 37°C mit ATP inkubieren; für komplexe Proben die Verdauung verlängern und das Enzym/ATP täglich nachfüllen, um die lineare Belastung zu reduzieren und eine Überverdauung zu vermeiden, die Kreise beschädigen könnte. Die Implementierungen von Circle-seq mit Plasmid-Safe sind zusammengefasst in Yu et al. (2023) Übersicht über Circle-seq.
Überprüfung ist nicht verhandelbar.
- qPCR von einem oder zwei Einzelkopie-genomischen Loci: Vergleichen Sie Ct vor und nach der Verdauung. Ein erheblicher Ct-Anstieg (z. B. ≥3–4 Zyklen) weist auf eine Depletion von linearer DNA hin. Mehrere Studien verwenden locus-spezifische Assays, um die Depletion zu quantifizieren und die Anreicherung zu bestätigen.
- Gel-Elektrophorese: Erwarten Sie das Verschwinden ausgeprägter linearer Bänder/Schmiere; zirkuläre Spike-ins sollten nach wie vor nachweisbar sein (siehe den Abschnitt zu den Kontrollen).
Mitochondriale DNA (mtDNA) Kontamination
- mtDNA kann bestehen bleiben und Ergebnisse verzerren. Viele Circle-Seq-Workflows linearisieren mtDNA vor der Verdauung mithilfe von CRISPR-Cas9 mit sgRNAs an zwei mtDNA-Stellen und bestätigen dann die Entfernung durch PCR; siehe Protokollhinweise in dos Santos et al. (2023).
QC-Prüfpunkt 2: Verdauungserfolg
- Dokumentieren Sie die Ct-Verschiebungen der qPCR. Erfassen Sie Gelbilder vor/nach der Verdauung.
- Wenn der Ct-Verschiebung <3 Zyklen beträgt und das Gel einen verbleibenden Schimmer zeigt, wiederholen Sie die Verdauung mit frischem Enzym und ATP. Achten Sie auf die Integrität des Kreises; vermeiden Sie längere Hochtemperaturexposition oder übermäßiges Pipettieren.
Entscheidungsregel
- Fahren Sie mit der RCA fort, nachdem Sie eine robuste lineare Depletion durch qPCR/Gel bestätigt haben. Wenn die Verdauung unvollständig bleibt, führen Sie den Vorgang mit erhöhten Enzymeinheiten, frischem ATP oder verlängerter Inkubation erneut durch. Wenn Sie vermuten, dass es zu einem Nicking des Kreises gekommen ist, wechseln Sie das Enzym oder reduzieren Sie die Expositionszeit.
Referenz- und tiefere Methodenauswahl
- Für Enzymwahl, Kompromisse und Kontrollstrategien siehe Auswahl von eccDNA-Anreicherungsmethoden.
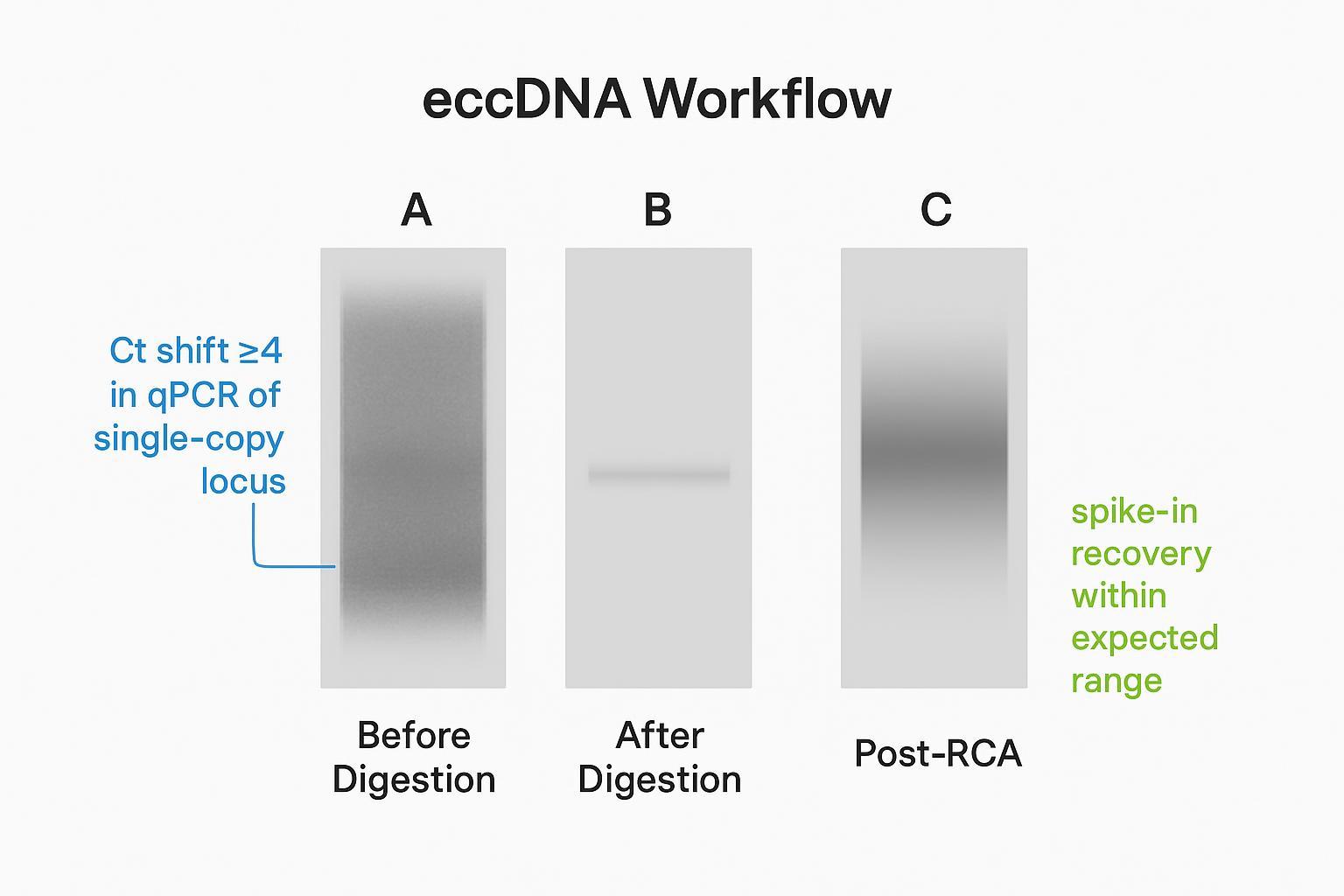 Abbildung 2. Gelvergleich: Vor der Verdauung — reichlich lineare Verwischung; Nach der Verdauung — erschöpfte lineare DNA mit erhaltenem zirkulären Spike-in (über qPCR überprüfen); Nach der RCA — Hochmolekularer Konkatemer-Verwischung, die auf eine erfolgreiche Amplifikation hinweist.
Abbildung 2. Gelvergleich: Vor der Verdauung — reichlich lineare Verwischung; Nach der Verdauung — erschöpfte lineare DNA mit erhaltenem zirkulären Spike-in (über qPCR überprüfen); Nach der RCA — Hochmolekularer Konkatemer-Verwischung, die auf eine erfolgreiche Amplifikation hinweist.
Schritt 3: Rolling Circle Amplifikation (RCA) mit Phi29
RCA verstärkt Kreise mit niedriger Abundanz, sodass sie in nachgelagerten Bibliotheken nachweisbar sind. Die Phi29-Polymerase ist das Arbeitstier aufgrund ihrer starken Strangverschiebung und hohen Prozessivität. Sie kann jedoch Konkatomere und Chimären erzeugen, wenn die Bedingungen nicht sorgfältig gesteuert werden.
Schlüsselparameter
- Primer: Verwenden Sie exonuklease-resistente zufällige Primer (z. B. phosphorothioatgeschützte), um die Integrität der Primer unter der Proofreading-Aktivität von Phi29 aufrechtzuerhalten; siehe Kit-Anleitung wie NEB's Phi29‑XT RCA-Kit-Protokoll.
- Temperatur und Dauer: WT phi29 läuft typischerweise bei ~30°C; ingenieurtechnisch modifizierte Varianten (z.B. phi29‑XT) bei 42°C. Die Begrenzung der RCA auf ~2 Stunden reduziert oft die übermäßige Bildung von Konkatmeren, während sie eine ausreichende Ausbeute liefert, wie in den Anwendungshinweisen verschiedener Anbieter für die multiple Displacements Amplifikation festgestellt wurde.
- Debranching: Die Behandlung mit T7 Endonuklease I oder sanftes akustisches Scheren kann verzweigte Konkatemere vor der Bibliotheksvorbereitung reduzieren. Die RCA-Richtlinien von NEB empfehlen das Debranching für sauberere Bibliotheken.
- Inaktivierung: Befolgen Sie die Kit-Protokolle für Wärme- oder EDTA-basierte Stopps, um die Aktivität zu stoppen.
Kontrollen in RCA
- Kein-Template-Kontrolle (NTC): sollte keine Amplifikation zeigen; jedes Signal weist auf Kontamination hin.
- Lineare negative Kontrolle: Führen Sie eine Scheinverdauung ohne zirkuläre Vorlagen durch; signifikante Amplifikation deutet auf unvollständigen Verdau oder Primerartefakte hin.
- Zirkuläre Spike-Ins: Quantifizierung des RCA-Ertrags und der Verzerrung über die Größen hinweg.
QC-Prüfpunkt 3: Nach-RCA-Profil
- Gel/TapeStation: Erwarten Sie ein hochmolekulares Schmiersignal. Deutliche wiederholte Banden können auf Concatemer-Artefakte hinweisen; mildern Sie dies durch verkürzte Reaktionszeit oder Debranching.
- Quantifizieren Sie den RCA-Ertrag; beachten Sie die Fragmentverteilung, wenn Sie eine Größenwahl planen.
Entscheidungsregel
- Wenn RCA übermäßig diskrete Bänder oder übermäßige Chimären produziert, verkürzen Sie die Inkubationszeit, überprüfen Sie die Primerqualität und ziehen Sie in Betracht, das Debranching vor der Fragmentierung durchzuführen. Bestätigen Sie, dass NTC und lineare Negativen sauber bleiben.
Kontext für Eingaben zu Replikationsstress
- Wenn Ihr Projekt absichtlich die Replikation stört (z. B. durch Hydroxyurea-Behandlungen), verstehen Sie, wie Stress die Häufigkeit von Zirkeln und die Eingaben für RCA beeinflusst. Erfahren Sie mehr in Replikationsstress und eccDNA.
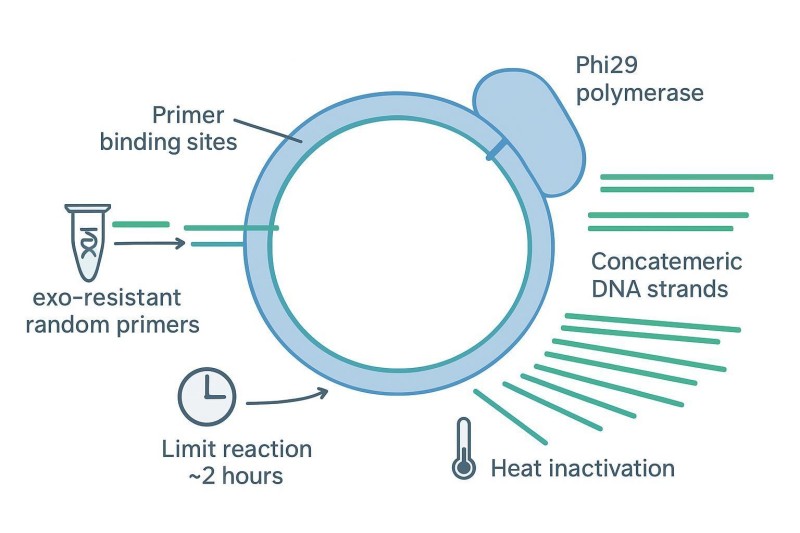 Abbildung 3. Phi29 RCA erzeugt concatemerische Amplicons aus zirkulären Vorlagen – Artefakte reduzieren durch Verkürzung der Reaktionszeit, Verwendung von exonuklease-resistenten Primern und Anwendung von Debranching oder sanftem Scheren vor der Bibliotheksvorbereitung.
Abbildung 3. Phi29 RCA erzeugt concatemerische Amplicons aus zirkulären Vorlagen – Artefakte reduzieren durch Verkürzung der Reaktionszeit, Verwendung von exonuklease-resistenten Primern und Anwendung von Debranching oder sanftem Scheren vor der Bibliotheksvorbereitung.
Schritt 4: Bibliotheksvorbereitung und Qualitätskontrolle
Mit angereicherten, amplifizierten zirkulären Vorlagen sind Sie bereit, Kurzlesebibliotheken (häufig Illumina PE150) für Entdeckung und Profiling vorzubereiten. Betrachten Sie die Bibliotheksvorbereitung sowohl als einen Konstruktionsschritt als auch als ein diagnostisches Verfahren.
Fragmentierung
- Zielinsertionslängen von etwa 300–400 bp. Überfragmentierung verringert die Komplexität und kann die Erkennung von Junctions verzerren.
- Wählen Sie akustisches Scheren oder enzymatische Fragmentierung; stimmen Sie Energie/Zeit ab, um das Insertfenster zu treffen.
Adapter-Ligation und Amplifikation
- Verwenden Sie Ligation-Kits, die mit Ihrer Fragmentierungsstrategie kompatibel sind; vermeiden Sie übermäßige PCR-Zyklen – jeder Zyklus erhöht Duplikationen und Verzerrungen. Wenn die Eingaben niedrig sind, ziehen Sie Ligation-Optionen in Betracht, die für DNA mit niedrigem Input optimiert sind.
Größenauswahl und Bereinigung
- SPRI-Perlenverhältnisse: Feinabstimmung, um Ihr Ziel-Insert-Fenster beizubehalten. Zu enge Auswahl kann informative, übergreifende Fragmente verwerfen.
Leitfaden zur Sequenzierungstiefe
- Discovery-Läufe: Viele Circle-Seq-Studien berichten von pro Probe Lesezahlen von mehreren zehn bis mehreren hundert Millionen PE150-Lesungen. Ihre optimale Tiefe hängt von der Gewebe-Komplexität und der erwarteten Kreis-Abundanz ab; beginnen Sie mit moderater Tiefe und skalieren Sie, sobald die Qualitätskontrolle bestanden ist. Für den Kontext der Plattform siehe die Überblick über Next-Generation Sequencing (Illumina).
- Validierungsdurchläufe: Wenn der Wechsel zur Long-Read-Bestätigung erfolgt (PacBio HiFi oder ONT), kann sich die Tiefe auf Kandidaten anstatt auf globale Entdeckung konzentrieren.
Akzeptanzkriterien zur Dokumentation
- Einfügen der Größenverteilung (Bioanalyzer/TapeStation) um 300–400 bp.
- Eine Mapping-Rate von nahe ~90% mit einem Standard-Aligner (z. B. BWA-MEM) weist auf eine gute Bibliotheksqualität hin.
- Duplikationsraten: Überwachen und so niedrig wie möglich halten; zusammen mit Q30-Metriken und Details zur Entfernung von Adaptern/niedriger Qualität berichten.
Wo ein Rundum-Service helfen kann
- Für Labore, die eine standardisierte Übergabe bevorzugen – von der Verdauung über die Bibliotheksvorbereitung bis hin zur Bioinformatik – Offenlegung: CD Genomics Unser Produkt bietet eccDNA-Sequenzierung und -Analyse an.
Referenzannahmekriterien
- Überprüfen Sie die detaillierten Akzeptanzkriterien, Reproduzierbarkeitsmetriken und Berichterstattungshinweise in Qualitätsmetriken für eccDNA-Sequenzierung.
Kontrollen und Spike-ins: Gestaltung einer End-to-End-Qualitätskontrolle
Der bedeutendste Faktor für die Reproduzierbarkeit in einem eccDNA-Sequenzierungs-Workflow ist der Kontrollplan. Integrieren Sie drei obligatorische Kontrollen in Ihre Pipeline und behandeln Sie deren Ausgaben als Projektwächter.
- Synthetische zirkuläre Spike-ins (kombiniert mit einer linearen Version)
- Was sie tun: die Effizienz der Verdauung und den RCA-Ertrag quantifizieren sowie die Nachweisgrenzen kalibrieren.
- So verwenden Sie es: Fügen Sie definierte Mengen eines zirkulären Plasmids oder Mini-Circles sowie eine linearisierte Version derselben Sequenz bei der Extraktion oder der Vorverdauung hinzu. Nach der Verdauung und RCA berechnen Sie das Log2-Verhältnis von zirkulär:linear durch qPCR oder Lesekonten.
- Was zu erwarten ist: Circle-seq-Studien berichten von erheblichen Erhöhungen der Verhältnisse von zirkulär zu linear nach optimierter Verdauung und RCA, was auf eine erfolgreiche lineare Depletion und zirkuläre Anreicherung hinweist; siehe Beispiele in Yu et al. (2023) Überblick über Circle-seq.
- Lineares genomisches DNA-Negativkontrolle
- Was es tut: Schätzt den Hintergrund von falsch-positiven Ergebnissen – falsche Junction-Anrufe oder Split-Reads, die nicht erscheinen sollten, wenn keine Kreise vorhanden sind.
- So verwenden Sie es: Verarbeiten Sie eine Probe durch Verdauung und RCA, die keine zirkulären Vorlagen enthält. Jegliche Amplifikation oder Junction-Erkennung weist auf unvollständige Verdauung oder RCA/Primer-Artefakte hin; verwenden Sie diesen Hintergrund, um die Erkennungsschwellen festzulegen.
- Keine-Vorlagen-RCA-Steuerung (NTC)
- Was es tut: überwacht Kontamination und Primerartefakte in RCA-Reagenzien.
- So verwenden Sie es: Führen Sie eine RCA mit Wasser anstelle von DNA durch. Jedes Produkt, das auf Kontamination hinweist – fahren Sie nicht fort.
eccDNA-Sequenzierungs-Workflow: QC-Schwellenwerte und Tiefenplanung
Erkennungsschwellen
- Akzeptanz von Kurzlesungen: Erfordern mehrere Split-Lesungen, die eine Junction unterstützen (z. B. ≥5 Split-Lesungen) und konsistente Abdeckung um den vermeintlichen Kreis. Passen Sie basierend auf dem Hintergrund an, der im linearen Negativkontrollversuch beobachtet wurde.
- Langzeitlese-Akzeptanz (für Kandidaten): Ziel sind mehrere unabhängige Voll-Längen-Lesungen, die die Junction abdecken (z. B. ≥2 ONT/PacBio-Lesungen), plus Split-Read-Bestätigung. Ansätze zur Validierung von Kandidaten mit vollständiger eccDNA-Rekonstruktion werden in Methoden wie FLED beschrieben; siehe Li et al. (2023) vollständige eccDNA-Erkennung.
Spike-in-Kalibrierung
- Verwenden Sie gemessene Wiederherstellung und Verhältnisänderungen, um die Sensitivität anzupassen. Wenn die Spike-in-Wiederherstellung schlecht ist, überprüfen Sie die Verdauung/RCA, bevor Sie tiefer sequenzieren.
Empfehlungen zur Sequenzierungstiefe
- Entdeckung: Beginnen Sie mit ~50–150 Millionen PE150-Reads pro Probe als pragmatischen Bereich und skalieren Sie dann entsprechend der Kontrollleistung und der Komplexität der Probe. Tiefenbereiche und Übergänge von Entdeckung zu Validierung werden in den eccDNA-Methodenpapieren diskutiert; eine allgemeine Übersicht über die Datenmengen von Circle-Seq erscheint in Jiang et al. (2023).
- Validierung: Für die Bestätigung gezielter Kandidaten können Langleseläufe bescheiden sein, wenn sie vollständige durchgehende Reads liefern; die Umfassender Überblick über Long-Read-Sequenzierung bietet Plattformkompromisse, die für die Tiefenplanung nützlich sind.
Aufzeichnung der QC-Ergebnisse
- Mindestens sollten erfasst werden: Änderungen des Spike-in-Verhältnisses und der Erholungsprozentsatz, qPCR Ct-Verschiebungen für Einzelkopieloci, Ergebnisse der NTC/Negativkontrollen, Verteilung der Bibliotheksinsertgrößen, Mapping-/Duplikationsraten und minimale unterstützende Reads für akzeptierte Zyklen.
Langzeit-Lesevalidierungsstrategie (PacBio/ONT)
Die Entdeckung von Kurzlesungen ist leistungsstark, aber komplexe Wiederholungen und Unklarheiten an den Verbindungsstellen profitieren von orthogonaler Validierung durch Langlesungen. Planen Sie diesen Schritt frühzeitig, damit Sie Material reservieren und eine erneute Extraktion vermeiden können.
Kandidatenauswahl
- Wählen Sie 5–10 vermeintliche eccDNAs aus, die unterschiedliche Größen und Wiederholungsinhalte abdecken. Bevorzugen Sie Kandidaten mit starker Unterstützung durch Kurzlesungen und guten Spike-in-Rückgewinnungsmetriken.
Bibliotheksstrategien
- Option A: Sequenz RCA-Konkatemer nativ auf ONT, um junction-überspannende Moleküle innerhalb langer tandemwiederholungen zu erfassen.
- Option B: Führen Sie PacBio HiFi-Bibliotheken auf größenselektierter DNA durch, um eine hohe Genauigkeit pro Lesevorgang zu erreichen; geeignet zur Bestätigung präziser Bruchstellen.
- Option C: hybrides Konzept – akustisches Scheren nach der RCA verwenden, um Längenverteilungen anzupassen, die für die gewählte Plattform geeignet sind.
Eingaben und Qualitätskontrolle
- Überprüfen Sie die Länge und Qualität der Bibliothek mit Bioanalyzer/TapeStation. Stellen Sie sicher, dass minimale Adapterdimer und eine angemessene Größenverteilung vorhanden sind.
- Bestätigen Sie, dass NTCs und lineare Negativen sauber bleiben; das Langzeit-Signal in Negativen deutet auf Kontamination oder Verdauungsfehler hin.
Interpretation von Langzeitdaten
- Suchen Sie nach vollständigen Lesevorgängen, die die Junction mit hoher Mapping-Qualität und minimalem Soft-Clipping durchqueren. Kombinieren Sie dies mit Split-Read-Beweisen aus Short-Read-Bibliotheken.
- Verwenden Sie graphbasierte Rekonstruktion oder Konsensassemblierung, um Wiederholungen zu lösen; erfordern Sie mehrere unabhängige Moleküle, bevor Sie die Struktur deklarieren.
Plattformkompromisse
- ONT ist hervorragend darin, sehr lange Konkatemer und komplexe Wiederholungen zu erfassen; PacBio HiFi bietet eine ausgezeichnete Genauigkeit pro Lesevorgang für hochzuverlässige Breakpoint-Aufrufe.
Wo der Service-Kontext hilft
- Wenn Sie einen schlüsselfertigen Validierungsweg benötigen, Umfassender Überblick über Long-Read-Sequenzierung erklärt Plattformkompromisse. Anbieter einschließlich CD Genomics Langzeit-Sequenzierung und PacBio SMRT-Sequenzierung Seitenumrissfähigkeiten, die Sie auf Ihren Validierungsplan abbilden können.
Häufige Fallstricke und Fehlersuche
Hoher linearer Hintergrund nach der Verdauung
- Wahrscheinliche Ursachen: unzureichende Enzymeinheiten, abgebautes ATP oder zu kurze Inkubationszeit.
- Korrekturen: Wiederholen der Verdauung mit frischem Enzym und ATP; Inkubation verlängern oder täglich nachdosieren (für Plasmid-Safe); mit qPCR Ct-Verschiebung und Gel verifizieren.
Mitochondriale Kontamination
- Wahrscheinliche Ursachen: mtDNA-Persistenz während der Verdauung.
- Korrekturen: mtDNA an zwei Stellen mit CRISPR-Cas9 vor der Verdauung prä-linearisieren; durch PCR bestätigen.
RCA-Kontaktemere und Chimären
- Wahrscheinliche Ursachen: verlängerte RCA, Primerabbau oder Verzweigung.
- Korrekturen: Begrenzen Sie RCA auf ~2 Stunden; verwenden Sie exo-resistente Primer; entzweigen Sie mit T7 Endonuklease I; stellen Sie sicher, dass NTC/lineare Negativen sauber sind. Anbieterprotokolle wie NEB's Phi29‑XT RCA-Protokoll Bitte stellen Sie Minderungshinweise zur Verfügung.
Überfragmentierung während der Bibliotheksvorbereitung
- Wahrscheinliche Ursachen: übermäßige Scherzeit oder Enzymexposition.
- Korrekturen: Fragmentierungsenergie/-zeit titrieren, um 300–400 bp Inserts zu erreichen; über Bioanalyzer überwachen; PCR-Zyklen reduzieren, um die Diversität zu erhalten.
Niedrige Bibliothekskomplexität oder hohe Duplizierung
- Wahrscheinliche Ursachen: niedrige Eingangs-DNA oder Überamplifikation.
- Korrekturen: Optimierung der Ligationseffizienz; Minimierung der PCR-Zyklen; Berücksichtigung von Anpassungen bei der Größenselektion; erneute Bewertung des RCA-Ertrags und der Qualitätskontrolle der Verdauung.
Bioinformatik Übergabe-Checkliste
Die Bereitstellung eines robusten Datensatzes für das Analyse-Team beschleunigt die Interpretation und reduziert den Austausch. Stellen Sie die folgenden Mindestanforderungen bereit:
- Rohlesetiefe (paired-end und Leselänge) pro Probe.
- Mapping-Rate, Duplikationsrate und Q30-Verteilung nach Adapter-/Qualitätsbeschnitt.
- Fügen Sie die Größenverteilung von Bioanalyzer/TapeStation ein.
- Spike-in-Wiederherstellungsmetriken: zirkuläre: lineare log2-Verhältnisänderungen und Prozentuale Wiederherstellung über Größen.
- qPCR Ct-Verschiebungen für Einzelkopien-Loci vor und nach der Verdauung.
- NTC und lineare negative Kontrollresultate (einschließlich aller erfassten Reads und Junction-Anrufe, idealerweise null).
- Erkannte Schwellenwerte verwendet: minimale Split-Reads pro Junction (Kurzlesung) und minimale Voll-Längen-Reads (Langlesung) für die Akzeptanz von Kandidaten.
- Eine Auswahlliste von Kandidaten, die für die Validierung von Langlesen ausgewählt wurden, mit Begründung.
Fazit: Standardisierung für reproduzierbare "circle-seq eccDNA"-Ergebnisse
Ein eccDNA-Sequenzierungsworkflow lebt oder stirbt durch seine Kontrollen. Sanfte HMW-Extraktion bewahrt die Kreise; rigorose Exonuklease-Digestion, verifiziert durch qPCR/Gel, entfernt den linearen Hintergrund; sorgfältig abgestimmte RCA amplifiziert die Kreise, ohne Ihre Bibliothek mit Ketten zu überfluten; und disziplinierte Bibliotheks-QC sowie sinnvolle Sequenzierungstiefe tragen zuverlässige Signale in die Analyse. Behandeln Sie synthetische zirkuläre Spike-ins, lineare negative Kontrollen und NTCs als obligatorisch. Planen Sie eine Langlesevalidierung für repräsentative Kandidaten, um Verbindungen und Wiederholungsstrukturen zu sichern.
Wenn Sie einen standardisierten, RUO-Weg von der Anreicherung über die Bibliotheksvorbereitung bis hin zur Berichterstattung wünschen, CD Genomics eccDNA-Sequenzierung kann das Projektdesign und die Ergebnisse unterstützen. Für einen tiefergehenden Kontext zur Plattform, besuchen Sie erneut die Überblick über Next-Generation Sequencing (Illumina) und das Umfassender Überblick über Long-Read-Sequenzierung.
Referenzen:
- Yu X, et al., "Circle‑seq Übersicht und Spike-in Quantifizierung" (2023) — Methoden und Spike-in Metriken: Yu et al., 2023 — Circle‑seq (PMC10788182).
- Li Y, et al., "Vollständige eccDNA-Detektion (FLED)" (2023) — Validierung mit langen Reads und Schwellenwerte für unterstützende Reads: Li et al., 2023 — FLED (PMC10632013).
- Jiang Z, et al., "Circle‑seq Tiefe und Entdeckungsüberlegungen" (2023) — Beispiele für Sequenzierungstiefe und Kontext des Datensatzes: Jiang et al., 2023 (PMC10300174).
- Hong J, et al., "HMW-Extraktion und eccDNA-Atlas" (2024) — Empfehlungen zur HMW-Extraktion und Kartierungsraten: Hong et al., 2024 (PMC10973517).
- Hansen K, et al., "Vergleich der Reinigung für eccDNA-Workflows" (2023) — auf Perlen basierende vs. alternative Reinigungen: Hansen et al., 2023 (PMC10523981).
- dos Santos R, et al., mtDNA-Linearisierungsprotokoll Notizen (2023) — CRISPR‑Cas9 Vor-Linearisierung Empfehlungen: dos Santos et al., 2023 (PMC10495552).
- Zhuang L, et al., "Kreisbewertung und Kontinuitätsmetriken" (2024) — Bewertungs-/Filtergrenzen und Kontinuitätsmetriken: Zhuang et al., 2024 (PMC10948907).
- Chen Q, et al., urinäre eccDNA-Beispiele (2025) — Anwendungs- und Nachweishinweise: Chen et al., 2025 (PMC12627897).
- Frontiers in Genetics Methodenübersicht (2024) — Überblick über die Auswahl von eccDNA-Extraktions- und Anreicherungsmethoden: Frontiers in Genetics Methodenübersicht (2024).
- Qiu et al., "Extrachromosomale zirkuläre DNA als neuartiger Biomarker" (PMC) — breiterer Kontext und Übersicht: Qiu et al. (Überblick) (PMC).
- Ye et al., PSD-Nutzung und methodische Hinweise (2023) — Beispielanwendungen von PSD in Circle-seq: Ye et al., 2023 (PMC10670553).
- Lin et al., PSD- und Anreicherungsbeschreibungen (2024) — zusätzliche PSD-Nutzungsbeispiele: Lin et al., 2024 (PMC11606223).
Autor
Yang H. — Leitender Wissenschaftler, CD Genomics; Universität Florida.
Yang ist ein Genomforschungswissenschaftler mit über 10 Jahren Forschungserfahrung in Genetik, molekularer und zellulärer Biologie, Sequenzierungsabläufen und bioinformatischer Analyse. Er ist sowohl in Laborverfahren als auch in der Dateninterpretation versiert und unterstützt das Design von RUO-Studien und NGS-basierten Projekten.


