
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
AAV-Genomsequenzierung
Was ist das AAV-Genom?
Das Genom des AAV (Adeno-assoziierten Virus) ist ein kleines, aber hochfunktionales einzelsträngiges DNA (ssDNA)-Molekül, das etwa 4,7 Kilobasen (kb) umfasst. Dieses kompakte genetische Paket ist geschickt mit mehreren wichtigen Merkmalen gestaltet:
- Inversierte terminale Wiederholungen (ITRs)An beiden Enden des Genoms gelegen, sind diese repetitiven Sequenzen entscheidend für den Lebenszyklus des Virus. Sie bilden charakteristische Haarnadelstrukturen, die für den Replikationsprozess und die Verpackung des viralen Genoms von wesentlicher Bedeutung sind. Diese ITRs spielen auch eine Schlüsselrolle bei der Integration der viralen DNA in das Genom der Wirtszelle.
- Rep-GeneDie Rep (Replikations-) Gene kodieren für Proteine, die für die Replikation des AAV-Genoms und die Regulierung seines Lebenszyklus unerlässlich sind. Diese Proteine sind an der Herstellung von Kopien der viralen DNA beteiligt und können auch dabei helfen, das genetische Material des Virus in das Genom des Wirts zu integrieren, um sicherzustellen, dass das Virus effektiv bestehen und sich verbreiten kann.
- Cap-GeneDie Cap-Gene produzieren die Proteine, die die schützende äußere Hülle des Virus, das Kapsid, bilden. Diese Kapsidproteine sind nicht nur strukturelle Komponenten; sie bestimmen die Fähigkeit des Virus, spezifische Zelltypen zu infizieren. Sie tragen auch zur Stabilität des Virus und seiner Effizienz bei, sein genetisches Material zu übertragen.
- Regulatorische RegionenDiese Regionen steuern die Expression der Rep- und Cap-Gene. Sie fungieren wie Verkehrsleiter und stellen sicher, dass die Produktion von Virusproteinen zur richtigen Zeit und in den richtigen Mengen während des Lebenszyklus des Virus erfolgt.
Was ist NGS-Sequenzierung von AAV?
NGS (Next Generation Sequencing)Next-Generation Sequencing) von AAV bietet einen tiefen Einblick in das virale Genom und enthüllt eine Fülle von Informationen über seine Struktur und potenzielle Probleme. Diese hochmoderne Technologie kartiert das gesamte AAV-Genom sorgfältig, identifiziert genetische Varianten, Mutationen und mögliche Verunreinigungen. Solche detaillierten Einblicke sind entscheidend für die Feinabstimmung von AAV-Vektoren, die in der Gentherapie verwendet werden, um sicherzustellen, dass sie sowohl sicher als auch effektiv sind. Darüber hinaus hilft NGS, die genaue Konzentration von AAV in Proben zu bestimmen und die Reinheit der Vektorpräparationen zu bestätigen, um sicherzustellen, dass sie frei von unerwünschten viralen oder bakteriellen Elementen sind. Im Wesentlichen ist NGS ein wichtiges Werkzeug, das die präzise Entwicklung, strenge Qualitätskontrolle und erfolgreiche Anwendung von AAV-basierten Gentherapien unterstützt.
Einführung in die AAV-Genomsequenzierung
AAVs sind derzeit eines der am häufigsten verwendeten und bedeutendsten Vektoren für die in vivo Gentherapie. Diese Vektoren können genetische Lasten von bis zu 4,7 kb liefern und können eine Vielzahl von Zelltypen mit minimaler Pathogenität infizieren. Die Produktion von AAVs beginnt mit dem Bau von Plasmiden und endet mit der Verpackung in Viruspartikel. Aufgrund der strukturellen Eigenschaften des AAV-Genoms ist die Herstellung von AAV-Viren mit hochpräzisem DNA inhärent herausfordernd. Um diese Komplexitäten zu bewältigen, bietet CD Genomics umfassende AAV-Genom-Sequenzierungs- und Qualitätskontrolllösungen an, die darauf ausgelegt sind, die Qualität der AAV-Vektoren zu optimieren.
Agarose-Gelelektrophorese, PCR/ddPCR und Southern Blotting wurden umfassend zur Bewertung der AAV-Genomgröße und zur Identifizierung spezifischer Regionen eingesetzt. Diese Methoden sind jedoch in ihrer Fähigkeit, umfassende Sequenzinformationen und Einblicke in die genomische Integrität zu liefern, eingeschränkt. Sequenzierungstechniken der dritten Generation, die für ihre Long-Read-Fähigkeiten bekannt sind, wurden als potenzielle Lösungen für die Analyse der Integrität des AAV-Genoms entwickelt. Dennoch erfordert die intrinsische Natur des AAV-Genoms als einzelsträngige DNA (ssDNA) dessen Umwandlung in doppelsträngige DNA vor der Bibliotheksvorbereitung für die Sequenzierung der dritten Generation.
Dieser Umwandlungsprozess, zusammen mit den modifizierten Termini in AAV-Genomen, erschwert die präzise Analyse der terminalen Sequenzen. Darüber hinaus zeigen fragmentierte AAV-Genome, die kürzer sind als ihre intakten Gegenstücke, während der Bibliotheksvorbereitung und Sequenzierung eine signifikante Verzerrung, was zu einer unverhältnismäßigen Repräsentation in Sequenzierungsdatensätzen führt. Folglich liefert die Analyse der Integrität von AAV-Genomen mit Hilfe von Sequenzierung der dritten Generation oft Ergebnisse, die die tatsächliche Integrität unterschätzen, was zu Datenverzerrungen führt. Diese Verzerrung hat schwerwiegende Auswirkungen auf nachfolgende Compliance-Prüfungen und den Arzneimittelzulassungsprozess. Daher ist die Sequenzierung der dritten Generation zwar für die genaue Sequenzanalyse von AAV-Genomen geeignet, jedoch unzureichend für die Bewertung der terminalen Sequenzen und der Gesamtgenomintegrität.
Angesichts der zunehmenden regulatorischen Anforderungen sind robuste Erkennungsprotokolle und analytische Methoden unerlässlich, um die Sicherheit und Konformität von AAVs zu gewährleisten. CD Genomics hat AAV-Genomsequenzierungs- und Qualitätskontrolldienste auf der Grundlage von Hochdurchsatz-Sequenzierungstechnologien eingeführt. Durch den Einsatz von Sequenzierung der zweiten Generation (NGS) und dritte Generation (Einzelmolekül-Echtzeit-SequenzierungDurch Sequenzierungsanalysen untersuchen wir die Reinheit von AAV, die Integrität der ITR-Sequenz und die Genauigkeit der Zielsequenz. Unsere Dienstleistungen sind darauf ausgerichtet, Kunden in der Entwicklung von Gentherapeutika dabei zu unterstützen, ihre AAV-Produktionsprozesse zu verbessern und somit die Sicherheit nachfolgender klinischer Studien zu gewährleisten.
Vorteile unseres AAV-Genom-Sequenzierungsdienstes
- Genau ErgebnisseDurch präzise Sequenzierungsmethoden erreichen wir eine vollständige Sequenzierung der ITR-Regionen und identifizieren effizient und genau Mutationsstellen innerhalb dieser Sequenzen.
- Schnelle SequenzierungUnser Ansatz stellt sicher, dass die Sequenzierung der ITR-Regionen zügig erfolgt, was zu verkürzten Bearbeitungszeiten für die Ergebnislieferung führt.
- Umfassende BeratungsdiensteCD Genomics bietet während des gesamten Projektlebenszyklus kontinuierliche technische Unterstützung, unabhängig davon, ob in der Anfangs-, Zwischen- oder Endphase.
- Compliance-gesteuerte ITR-SequenzierungCD Genomics betreibt voll akkreditierte Labore, die mit modernsten Instrumenten ausgestattet sind und strikt an Qualitätsmanagementkontrollsysteme sowie standardisierte Betriebsverfahren (SOPs) halten.
Anwendungen der AAV-Genomsequenzierung
Gentherapieforschung
- Optimierung von AAV-Vektoren
- Bewertung der therapeutischen Ergebnisse
Genom-Editierung
- Effizienzanalyse
- Spezifitätsvalidierung
Impfstoffentwicklung
- Lieferungssystemoptimierung
- Immunogenitätsbewertung
Grundlagenforschung
- Erforschung des Infektionsmechanismus
- Auswirkungen von Virusvarianten
AAV-Genom-Sequenzierungs-Workflow
Unser hochqualifiziertes Expertenteam führt das Qualitätsmanagement nach jedem Verfahren durch, um umfassende und genaue Ergebnisse sicherzustellen. Unser AAV-Sequenzierungsworkflow ist unten aufgeführt, einschließlich Bibliotheksvorbereitung, Sequenzierung und bioinformatischer Analyse.

Dienstspezifikationen
Musteranforderungen
|
|
 Klicken |
Sequenzierungsstrategie
|
 |
Bioinformatikanalyse
Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an:
|
Analyse-Pipeline

Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details zur AAV-Genomsequenzierung für Ihre Schreibanpassung.
CD Genomics verwendet die Kurzlese-Ganzgenomsequenzierung, um die AAV-Genomsequenzierung durchzuführen, und integriert Sanger-Sequenzierung und Hochdurchsatzplattformen für umfassende Bewertungen. Dieser Ansatz ermöglicht die Untersuchung der Genomintegrität, die Erkennung von Heterogenität und die Identifizierung von DNA-Verunreinigungen in AAV-Bibliotheksvorbereitungen. Wir sind bestrebt, unser umfangreiches Fachwissen und unsere modernste Technologie zu nutzen, um überlegene Dienstleistungen und hochwertige Produkte anzubieten, die auf die spezifischen Bedürfnisse unserer Kunden zugeschnitten sind.
Demo-Ergebnisse
Teilweise Ergebnisse sind unten aufgeführt:
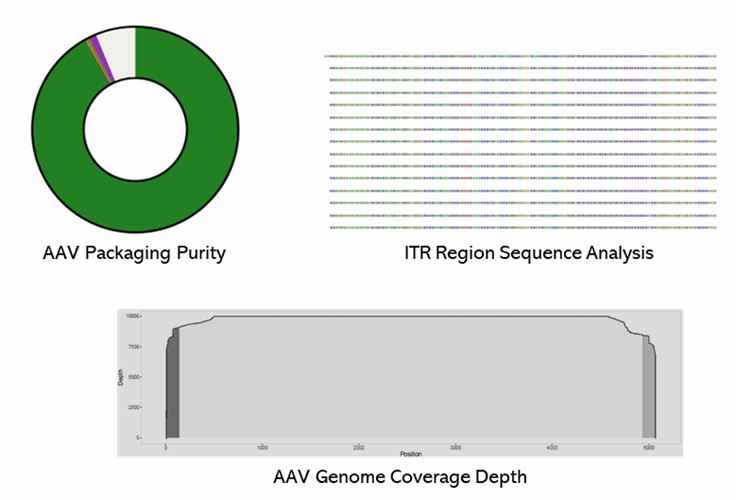
AAV-Genom-Sequenz-FAQs
1. Warum AAV für die Gentherapie wählen?
AAV zeichnet sich in der Gentherapie durch sein hohes Sicherheitsprofil, seine geringe Immunogenität, physikalische Stabilität, breites zelluläres Tropismus und verlängerte in vivo-Expression aus. Diese Eigenschaften machen AAV zu einem der am häufigsten verwendeten Vektoren in diesem Bereich. Der Bau von AAV-basierten Vektoren ist ein Grundpfeiler der Gentherapietechnologie.
Forscher haben umfangreiche Studien durchgeführt, um AAV-Vektoren zu optimieren. Diese Bemühungen umfassen die Modifizierung der genomischen Struktur des Virusvektors, die Erhöhung seiner Transportkapazität, die Steigerung des viralen Ertrags und die Verbesserung der Transduktionseffizienz in verschiedenen Gewebetypen. Solche Modifikationen zielen darauf ab, AAV-Vektoren an die spezifischen Bedürfnisse verschiedener Krankheiten anzupassen.
2. Warum sind die Kosten für AAV so hoch?
- Reinheitsanforderungen für AAV-Vektoren
AAV-Vektoren müssen strengen Reinheitsstandards genügen. Leere virale Kapsiden können Immunantworten auslösen und mit vollständig verkapselten Vektoren um die Infektion von Patienten-Zellen konkurrieren, was höhere Dosen erforderlich macht, um therapeutische Wirksamkeit zu erzielen. Diese Erhöhung der Dosis erhöht nicht nur das Risiko von Nebenwirkungen bei hohen Dosen, sondern steigert auch die potenzielle Hepatotoxizität und Karzinogenität. Folglich wird die Durchführung einer gründlichen Reinheitsvalidierung nach dem Bau von AAV-Vektoren zu einem wesentlichen Schritt.
- Hohe Mutationsraten in spezifischen ITR-Strukturen
Das AAV-Genom ist an beiden Enden von inversen terminalen Wiederholungsequenzen (ITRs) flankiert, die entscheidend für die Verpackung und Replikation von rAAV sind. Die ITR-Regionen in AAV-Plasmiden sind jedoch aufgrund ihrer palindromischen Sequenzen und des hohen GC-Gehalts instabil, was sie während der bakteriellen Replikation anfällig für Mutationen oder Deletionen macht. Selbst bei den standardisiertesten experimentellen Protokollen können AAV-Plasmide Mutationsraten von 5 % bis 15 % aufweisen.
- Herausforderungen bei der ITR-Erkennung
Angesichts der hohen Frequenz von ITR-Mutationen ist es entscheidend, die Integrität der ITR-Regionen in Plasmiden vor der viralen Verpackung zu bewerten. Diese Regionen sind jedoch reich an palindromischen Sequenzen, die in der Lage sind, stabile sekundäre Strukturen zu bilden. Während der Sanger-Sequenzierung können diese stabilen Haarnadel-Schleifen die PCR-Amplifikation hemmen, was zu Unterbrechungen bei der Sequenzierung führt und die Validierung der Sequenz erschwert. Ähnlich kann die konventionelle Methode mit Restriktionsenzymen unter Verwendung von SmaI zur Überprüfung der ITR-Integrität unzureichend sein, da kleinere Deletionen möglicherweise durch Gelelektrophorese unentdeckt bleiben.
3. Was sind die Vorteile der Verwendung von AAV als Vektor für die Gentherapie?
AAV hat mehrere Vorteile, darunter eine geringe Immunogenität, eine breite Wirtspalette, stabile physikochemische Eigenschaften und eine langfristige Expression exogener Gene.
Konkret sind diese Vorteile:
- Hohes SicherheitsprofilAAV ist im Allgemeinen nicht pathogen für Menschen oder verursacht nur sehr milde Symptome.
- Gezielte IntegrationAAV kann spezifisch in die AAVS1-Stelle auf dem menschlichen Chromosom 19 integrieren, was das Risiko von insertioneller Mutagenese verringert, das mit der zufälligen Integration anderer viraler Vektoren verbunden ist.
- Nachhaltige GenexpressionAAV-vermittelte Gentransfersysteme ermöglichen eine anhaltende und stabile Expression der eingeführten Gene, die durch umgebende genetische Elemente reguliert werden kann.
- Breite Host-ReichweiteAAV kann eine Vielzahl von Zelltypen infizieren, einschließlich sowohl teilender als auch nicht teilender Zellen.
- Thermische und chemische StabilitätDas AAV-Transfer-System zeigt eine signifikante thermische Stabilität und Widerstandsfähigkeit gegenüber sauren und alkalischen Bedingungen sowie organischen Lösungsmitteln, was es praktisch für die Lagerung macht.
AAV-Genom-Sequenzierungs-Fallstudien
Die Sequenzierung der Genompopulation von adenoassoziierten Viren erreicht eine vollständige Vektorgenomauflösung und enthüllt Mensch-Vektor-Chimären.
Journal: Molekulare Therapie Methoden & Klinische Entwicklung
Impactfaktor: 4,771
Veröffentlicht: 27. Februar 2018
Hintergrund
Rekombinante adeno-assozierte Viren (rAAVs) erfordern eine präzise Qualitätskontrolle für die Gentherapie. Traditionelle Methoden wie qPCR und Gelelektrophorese erfassen nicht vollständig die Genomfragmentierung oder Kontamination. Einzelmolekül-Echtzeit-Sequenzierung (SMRT) bietet einen detaillierten Überblick über sowohl vollwertige als auch verkürzte rAAV-Genome und zeigt Fehler bei der Genomverpackung sowie Kontaminationen durch Wirtszell- oder Virusfragmente auf. Diese fortschrittliche Technik ermöglicht eine umfassende Analyse der rAAV-Vektorpopulationen.
Materialien & Methoden
Probenvorbereitung
- Reinige virale Vektoren
- DNA-Extraktion
Sequenzierung
- Bibliotheksvorbereitung
- SMRT-Sequenzierung
- Pacific Biosciences RSII
- Leseverarbeitung
- Ausrichtung
- Circos-Diagramme, Venn-Diagramm-Visualisierung
- Lesen-Zähl-Normalisierung
Ergebnisse
AAV-GPseq verwendet SMRT-Sequenzierung, um vollständige scAAV-Genome von 5' bis 3' ITRs zu analysieren. Es zeigt, dass shRNA-Kassetten zu kürzeren Genomen führen und ermöglicht eine detaillierte Charakterisierung der ITR-Orientierungen sowie der Plus-/Minus-Strang-Verhältnisse, was Einblicke in die Verpackung und Replikationsdynamik von AAV-Vektoren bietet.
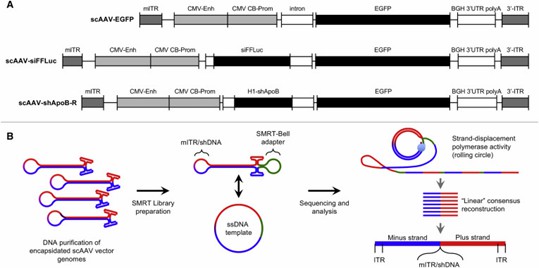 Abbildung 1. Einzelpartikel-Auflösungsprofilierung von scAAV-Genomen durch AAV-Gpseq.
Abbildung 1. Einzelpartikel-Auflösungsprofilierung von scAAV-Genomen durch AAV-Gpseq.
AAV-GPseq zeigt, dass die meisten Vektorgenom-Lesungen kürzer als 500 bp sind, was von der Gelanalyse abweicht. Durch die Verwendung von Spike-in-DNA zur Normalisierung quantifiziert es genau Voll-Längen- gegenüber truncierten Genomen. Diese Methode zeigt, dass Voll-Längen-Vektoren weniger verbreitet sind, als es die traditionellen Gel-Ergebnisse vermuten lassen.
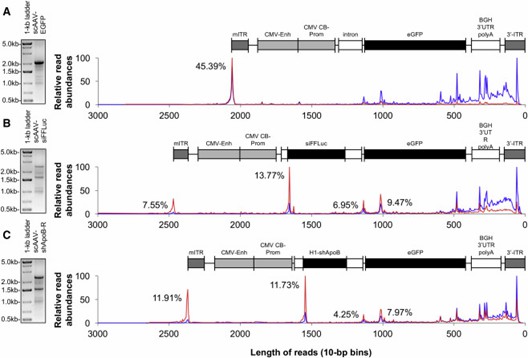 Abbildung 2. Einschätzung der Häufigkeit heterogener AAV-Genom-Populationen.
Abbildung 2. Einschätzung der Häufigkeit heterogener AAV-Genom-Populationen.
AAV-GPseq erkennt Plasmid-Rückgratsequenzen in Vektorgenomen, indem es Reads offenbart, die über die ITR-Regionen hinausgehen. Diese Methode identifiziert Reads, die mit Plasmid-Rückgratsequenzen übereinstimmen, was auf das Vorhandensein von umgekehrt verpackten oder größeren als einheitlichen Genomen hindeutet. Die Analyse bestätigt auch, dass viele kurze Genome unter 500 bp ITR-Sequenzen enthalten, aber in gereinigten Vektormustern nicht in nachweisbaren Mengen vorhanden sind.
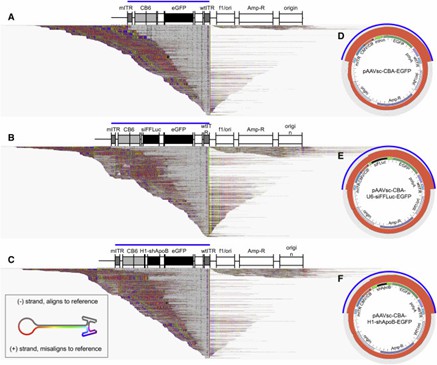 Abbildung 3. Ausrichtungen heterogener Vektorpopulationen auf das pCis-Plasmid-Referenz.
Abbildung 3. Ausrichtungen heterogener Vektorpopulationen auf das pCis-Plasmid-Referenz.
Fazit
Traditionelle Methoden zur Bewertung der Integrität von rAAV-Vektoren sind in der Profilierung einzelner Genome eingeschränkt. AAV-GPseq verbessert dies, indem es Genome von ITR zu ITR analysiert und chimäre Sequenzen sowie Plasmid-Rückgrate aufdeckt. Trotz seiner Stärken gibt es Herausforderungen, darunter die Unterrepräsentation längerer chimärer Sequenzen und Einschränkungen bei einzelsträngigen AAVs. Insgesamt verbessert AAV-GPseq die Qualitäts- und Sicherheitsbewertung von Gentherapie-Vektoren.
Referenz
- Tai P W L, Xie J, Fong K, et al. Die Sequenzierung der Genompopulation von adenoassoziierten Viren erreicht eine vollständige Auflösung des Vektorgenoms und zeigt menschlich-Vektor-Chimären. Molekulare Therapie-Methoden & Klinische Entwicklung, 2018, 9: 130-141.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Die HLA-Klasse-I-Immunopeptidome der AAV-Kapsidproteine
Zeitschrift: Frontiers in Immunologie
Jahr: 2023
Isolation und Charakterisierung neuer menschlicher Trägerpeptide aus zwei wichtigen Impfstoff-Immunogenen
Journal: Impfstoff
Jahr: 2020
Änderung des Gewichts, des BMI und der Körperzusammensetzung in einer bevölkerungsbasierten Intervention im Vergleich zu einer genetisch basierten Intervention: Die NOW-Studie
Journal: Fettleibigkeit
Jahr: 2020
Sarecyclin hemmt die Proteintranslation im Cutibacterium acnes 70S-Ribosom durch einen Zwei-Stellen-Mechanismus.
Zeitschrift: Nucleic Acids Research
Jahr: 2023
Identifizierung eines Darmkommensalen, der die blutdrucksenkende Wirkung von Ester-Angiotensin-Converting-Enzym-Hemmern beeinträchtigt.
Zeitschrift: Hypertonie
Jahr: 2022
Eine Splice-Variante im SLC16A8-Gen führt zu einem Defizit beim Laktattransport in aus menschlichen iPS-Zellen abgeleiteten retinalen Pigmentepithelzellen.
Zeitschrift: Zellen
Jahr: 2021
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


