
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Was ist DRUG-seq?
DRUG-seq, oder digitale RNA-Perturbation von Genen-Sequenzierung, ist eine hochmoderne RNA-Sequenzierungstechnologie, die hochgradige Arzneimittel-Screenings und eingehende Wirkmechanismusstudien ermöglicht. Im Gegensatz zu traditionellen RNA-seq-Methoden, die große Mengen an Ausgangsmaterial und komplexe RNA-Extraktionsschritte erfordern, liefert DRUG-seq umfassende Transkriptom-Profilierungen direkt aus Zelllysaten, Organoiden oder Gewebeschnitten – selbst wenn die Probenmengen extrem begrenzt sind.
Mit DRUG-seq können Forscher Hunderte bis Tausende von Arzneimittelbehandlungen oder genetischen Störungen parallel analysieren, Genexpressionssignaturen aufdecken, neuartige Biomarker entdecken und komplexe Wege offenbaren, die von therapeutischen Verbindungen beeinflusst werden. Ob für die Arzneimittelentdeckung, Toxizitätstests oder die Entwicklung personalisierter Medizin, DRUG-seq bietet die Skalierbarkeit, Geschwindigkeit und Präzision, die in der modernen Forschung erforderlich sind.
Unsere DRUG-seq-Service-Pakete
Bei CD Genomics verstehen wir, dass jedes Forschungsprojekt einzigartig ist. Deshalb bieten wir unsere Drug-seq-Dienste als flexible, modulare Pakete an, die auf Ihre wissenschaftlichen Ziele, Ihr Budget und Ihre Probenbeschränkungen abgestimmt sind.

Standard DRUG-seq
- 3' RNA-Seq konzentriert sich auf die Quantifizierung der Genexpression
- Geeignet für das Screening großer Bibliotheken von Verbindungen
- Unterstützt 96- oder 384-Well-Plattenformate
- Kosteneffiziente Lösung für Hochdurchsatzbedürfnisse
- Anwendungen:
- Wirkmechanismus-Studien
- Phänotypische Screening
- Toxizitätsbewertungen
DRUG-seq2
- Ultra-niedrige Eingabe, beginnend bei nur 1.000 Zellen
- Perfekt für Organoide, Gewebeproben oder seltene klinische Proben.
- Direkt-aus-Lysat-Workflow – keine RNA-Extraktion erforderlich
- Bietet höhere Empfindlichkeit und schnellere Bearbeitungszeiten.
- Breite Anwendungen:
- Tumorgewebsursprungstracing
- Entdeckung von Biomarkern
- Einblicke in die personalisierte Medizin
Vollständige DRUG-seq
- Umfassende vollständige Transkriptabdeckung
- Erkennt Spleißvarianten, Fusionstranskripte und isoformspezifische Arzneimittelreaktionen.
- Empfohlen für Projekte, die tiefere molekulare Einblicke erfordern.
- Eine höhere Sequenzierungstiefe (5–20 Millionen Reads pro Probe) gewährleistet robuste Ergebnisse.
- Ideal für:
- Analyse des alternativen Spleißens
- Detaillierte MOA-Erforschung
- Komplexe biologische Systeme und Organoidstudien
Warum sollten Sie sich für unsere DRUG-seq CRO-Dienstleistungen entscheiden?
- Keine RNA-Extraktion erforderlich
Optimieren Sie Ihren Arbeitsablauf und bewahren Sie die Integrität der Proben mit der direkten Bibliotheksvorbereitung aus Lysat. - Ultra-Niedriges Proben-Eingang
Analysieren Sie knappe oder wertvolle Proben – einschließlich einzelner Organoide oder kleiner Gewebeschnitte – mit nur 1.000 Zellen. - Skalierbare Hochdurchsatzlösungen
Screenen Sie Hunderte oder Tausende von Verbindungen gleichzeitig, um Zeit und Kosten für großangelegte Studien zu reduzieren. - Schnelle Bearbeitungszeiten
Erhalten Sie hochwertige Daten in nur 10 Geschäftstagen für standardmäßige DRUG-seq und in drei Wochen für vollständige Transkriptomik. - Regulatorische Qualitätskontrolle
Wir halten strenge Qualitätskontrollstandards ein, um die Zuverlässigkeit und Reproduzierbarkeit Ihrer Ergebnisse sicherzustellen. - Engagierte Bioinformatik-Expertise
Unsere Bioinformatiker bieten sowohl Standardanalysen als auch maßgeschneiderte Lösungen, die auf Ihre experimentellen Bedürfnisse abgestimmt sind. - US- und internationale Operationen
Mit Einrichtungen in den USA und Partnerschaften weltweit bieten wir globale Unterstützung und lokalen Service.

Wichtige Anwendungen von DRUG-seq-Diensten
Studien zum Wirkmechanismus von Arzneimitteln (MOA)
- Identifizieren Sie, wie Medikamente die Genexpressionswege beeinflussen.
- Off-Target-Effekte und sekundäre Mechanismen aufdecken
- Vergleichen Sie die Transkriptionsprofile über Verbindungenbibliotheken hinweg.
CRISPR-Störung-Profilierung
- Studie der kombinierten Effekte von genetischen Veränderungen und Medikamentenbehandlungen
- Bestimmen Sie synergistische oder antagonistische Wechselwirkungen auf transkriptomischer Ebene.
- Beschleunigen Sie die funktionelle Genomforschung zur Validierung von Arzneimittelzielen.
Tumorgewebe-Ursprungsverfolgung
- Verwenden Sie transkriptomische Signaturen, um Tumore nach ihrem Ursprung zu klassifizieren.
- Informieren Sie diagnostische Strategien und personalisierte Behandlungsentscheidungen.
- Analysieren Sie knappe Biopsieproben mit Protokollen für ultra-niedrigen Input.
Biomarker-Entdeckung
- Molekulare Marker zur Vorhersage der Arzneimittelreaktion erkennen
- Unterstützung der Entwicklung von personalisierter Medizin und gezielter Therapie
- Integrieren Sie transkriptomische Daten in Multi-Omics-Workflows.
Organoid- und Zellkultur-Optimierung
- Optimierung der Wachstumsbedingungen für komplexe Zellmodelle
- Identifizieren Sie Verbindungen, die die Lebensfähigkeit oder Differenzierung von Organoiden fördern.
- Verbessern Sie die Reproduzierbarkeit und Übersetzbarkeit von In-vitro-Studien.
Von Hochdurchsatz-Screening bis hin zu Durchbrüchen in der Präzisionsmedizin ermöglichen unsere DRUG-seq CRO-Dienste Ihre Arzneimittel-Seq-Analyseprojekte mit unvergleichlicher Skalierbarkeit und Tiefe.
Technische Spezifikationen
| Spezifikation | Einzelheiten |
|---|---|
| Unterstützte Formate | 96-Well-Platten 384-Well-Platten 1536-Well-Platten (für großangelegte Studien) |
| RNA-Extraktion | Nicht erforderlich – direkte Arbeitsabläufe aus Lysat |
| Sequenzierungsplattformen | Illumina und Nanopore |
| Sequenzierungstiefe (Standard / DRUG-seq2) | ~1 Million Reads pro Probe (typisch) |
| Sequenzierungstiefe (Vollständige DRUG-seq) | 5–20 Millionen Reads pro Probe für umfassende Abdeckung |
| Datenlieferungen | FASTQ-Dateien Ausrichtungsberichte Genanzahlmatrizen Optionale erweiterte Analyse |
DRUG-seq Dienstablauf
Schritt 1 – Probenvorbereitung
- Bereiten Sie Zellen, Organoide oder Gewebe-Lysate gemäß unseren detaillierten Einreichungsrichtlinien vor.
- Keine RNA-Extraktion erforderlich – direkt von Zelllysaten fortfahren.
Schritt 2 – Musterlieferung
- Versenden Sie gefrorene Platten oder Lysate an unser ausgewähltes Servicezentrum.
- Unser Team steht zur Verfügung, um Sie über die richtigen Verpackungs- und Versandbedingungen zu beraten.
Schritt 3 – Bibliotheksvorbereitung
- Weisen Sie jeder Probe eindeutige Barcodes zu.
- Erstellen Sie Sequenzierungsbibliotheken mit unseren optimierten DRUG-seq- oder Full-Length DRUG-seq-Protokollen.
Schritt 4 – Qualitätskontrollpunkt
- Bewerten Sie die Bibliotheksqualität mit Qubit-Assays, Fragmentanalysator und flacher Sequenzierung.
- Berichten Sie die QC-Ergebnisse zur Genehmigung durch den Kunden vor der Tiefensequenzierung.
Schritt 5 – Tiefe Sequenzierung
- Führen Sie Hochdurchsatz-Sequenzierung auf Illumina NovaSeq oder anderen kompatiblen Plattformen durch.
- Sequenzierungstiefe, die auf Ihre experimentellen Ziele zugeschnitten ist.
Schritt 6 – Datenanalyse & Berichterstattung
- Richten Sie die Reads an Ihrem gewünschten Genom aus.
- Generieren Sie Genzählmatrizen, QC-Berichte und optionale Analysen der differentiellen Genexpression.
Schritt 7 – Datenlieferung
Liefern Sie alle Daten sicher, einschließlich:
- FASTQ-Dateien
- Ausrichtungszusammenfassungen
- Genanzahlmatrizen
- Optionale benutzerdefinierte Analysen
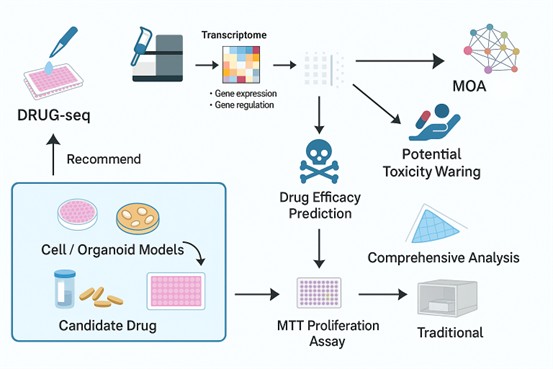
Von der initialen Probenvorbereitung bis zur umfassenden bioinformatischen Analyse bietet CD Genomics einen zuverlässigen, transparenten Workflow, der Ihre Forschungszeitpläne in den Vordergrund stellt.
Bioinformatikanalyse

Beispielanforderungen
| Servicepaket | Empfohlene Zelleneingabe (pro Well) | Notizen |
|---|---|---|
| Standard DRUG-seq | 2.000 – 20.000 Zellen | Geeignet für großangelegte Verbindungenstests |
| DRUG-seq2 | Schon mit nur 1.000 Zellen | Ideal für Organoide, Gewebeproben oder seltene klinische Proben |
| Vollständige DRUG-seq (96-Well) | 5.000 – 25.000 Zellen | Mindestens ~80.000 Zellen insgesamt pro Pool werden für optimale Qualität empfohlen. |
| Vollständige DRUG-seq (384-Well) | 2.000 – 10.000 Zellen | Das Gleiche wie oben |
| Probenart | Zellen, Organoide, Gewebelysate | Kontaktieren Sie uns für Handhabungsempfehlungen. |
Demo-Ergebnisse
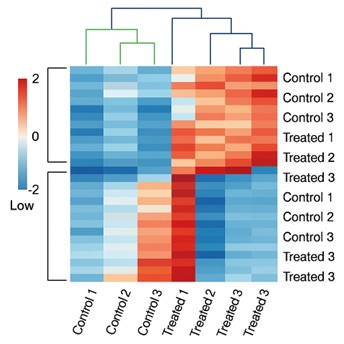
Genexpressions-Hitzekarte
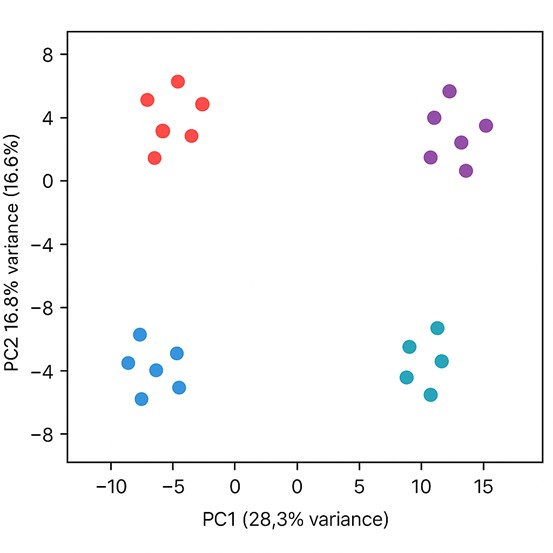
PCA-Diagramm
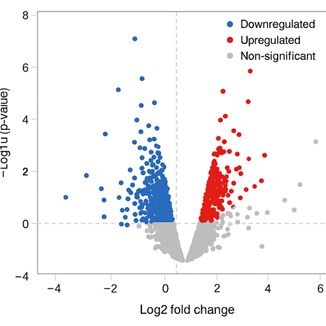
Vulkan-Diagramm
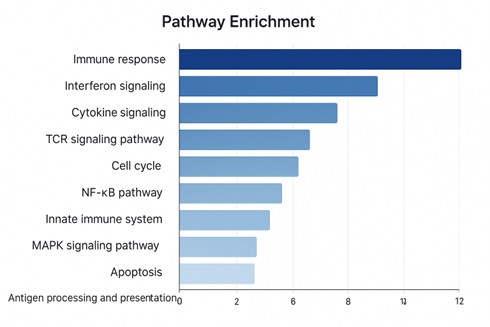
Wegbereicherung Säulendiagramm
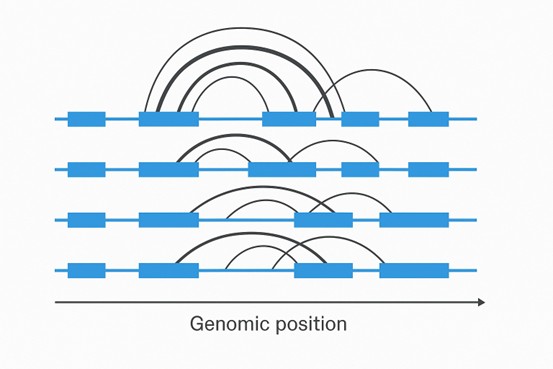
Spleißung / Isoform-Diagramm (Nur Vollständige DRUG-seq)
Drug-seq Dienstleistungs-FAQs
1. Was ist die minimale Zellzahl, die für DRUG-seq erforderlich ist?
Für das standardmäßige DRUG-seq empfehlen wir, mit mindestens 2.000–20.000 Zellen pro Vertiefung zu beginnen. Unser DRUG-seq2-Service kann jedoch mit so wenigen wie 1.000 Zellen pro Vertiefung arbeiten, was ihn ideal für begrenzte oder wertvolle Proben wie Organoide oder Gewebeschnitte macht.
2. Ist die RNA-Extraktion vor der Sequenzierung notwendig?
Nein. Einer der größten Vorteile von DRUG-seq ist, dass keine RNA-Extraktion erforderlich ist. Die Bibliotheken werden direkt aus Zelllysaten hergestellt, was die Arbeitsabläufe vereinfacht und die RNA-Integrität bewahrt.
3. Kann DRUG-seq Organoid-Proben analysieren?
Absolut. Unsere DRUG-seq und DRUG-seq2 Dienstleistungen sind sehr kompatibel mit Organoid-Modellen und anderen komplexen 3D-Zellsystemen, selbst bei extrem niedrigen Eingabemengen.
4. Wie lange dauert in der Regel die Bearbeitung von DRUG-seq-Diensten?
Für Standard-DRUG-seq-Projekte können die Daten in nur 10 Werktagen geliefert werden. Vollständige DRUG-seq-Projekte, die eine umfassendere Transkriptanalyse beinhalten, benötigen in der Regel etwa drei Wochen.
5. Welche Arten von Daten erhalte ich von DRUG-seq?
Standardliefergegenstände umfassen:
- Rohdaten im FASTQ-Format
- Ausrichtungsberichte
- Genanzahlmatrizen
- Optionale Berichte zur differentiellen Genexpression und Pfadanalyse
6. Welche Sequenzierungstiefe wird für DRUG-seq-Projekte empfohlen?
- Standard DRUG-seq / DRUG-seq2: ~1 Gb pro Well wird für eine zuverlässige Genexpressionsprofilierung empfohlen.
- Vollständige DRUG-seq: 5–20 Millionen Reads pro Probe werden für eine umfassende Transkriptabdeckung und Isoformanalyse empfohlen.
7. Beeinflusst die Zelllysezeit die cDNA-Qualität?
Ja. Übermäßig lange Lysezeiten können zu RNA-Abbau und führen zu einem höheren Anteil an kleinen Fragmenten in der cDNA-Bibliothek. Befolgen Sie immer die empfohlenen Inkubationszeiten, um qualitativ hochwertige Daten sicherzustellen.
8. Wie identifiziert DRUG-seq einzelne Proben in gepoolten Sequenzierungsdurchläufen?
Jeder Brunnen ist einzigartig mit einem beschriftet. gut Barcode und einen molekularen Barcode während der cDNA-Synthese. Nach dem Pooling und der Sequenzierung verwenden bioinformatische Pipelines diese Barcodes, um die Daten genau zu demultiplexen und die Genexpressionsprofile für jede Probe wiederherzustellen.
9. Kann dasselbe Wells-Barcodes und molekulare Barcodes für mehrere Experimente wiederverwendet werden?
Nein. Jede Kombination von molekularen Barcodes ist für Einwegartikel um Kreuzkontamination zwischen Proben zu verhindern. Handhaben Sie Barcodes immer gemäß den Protokollanweisungen, einschließlich des Auftauens auf Eis und des ordnungsgemäßen Mischens.
10. Für welche Anwendungen ist DRUG-seq geeignet?
- Studien zum Wirkmechanismus von Arzneimitteln
- CRISPR-Störung-Profilierung
- Tumorgewebe-Ursprungsverfolgung
- Entdeckung von Biomarkern
- Toxizitätsscreening
- Optimierung von Organoid-Modellen
11. Können Sie maßgeschneiderte bioinformatische Analysen anbieten?
Ja. Unser Bioinformatik-Team bietet sowohl standardisierte als auch vollständig maßgeschneiderte Datenanalyse-Lösungen an, um Ihre wissenschaftlichen Ziele zu erreichen.
12. Ist DRUG-seq kosteneffektiv für große Screenings?
Ja. Die hohe Multiplexing-Kapazität von DRUG-seq reduziert die Kosten pro Probe erheblich, was es zu einer ausgezeichneten Wahl für Hochdurchsatz-Verbindungsscreenings und großangelegte Transkriptomik-Projekte macht.
Drug-seq Dienstfallstudien
Fallstudie: Hochdurchsatz-MoA-Profiling von 433 Verbindungen mittels DRUG-seq
Ye, C. et al."DRUG-seq für miniaturisierte Hochdurchsatz-Transkriptom-Profilierung in der Arzneimittelentdeckung." Naturwissenschaftliche Kommunikation 9, 4307 (2018).
Methode und technischer Ansatz
- Hochdurchsatz-Multiplexing: Genutzte DRUG-seq-Plattform in 384- und 1536-Well-Formaten, die eine gleichzeitige Profilierung von 433 Verbindungen über 8 Dosisstufen ermöglicht.
- Barcode- und UMI-Kennzeichnung: Frühes Barcoding während der reversen Transkription, Pooling von Proben nach der RT, wodurch die Notwendigkeit einer individuellen RNA-Extraktion entfällt.
- Sequenzierungstiefe: Verwendete 1–2 Millionen Reads pro Well – ausreichend, um ~11.000 Gene pro Probe zu quantifizieren, im Vergleich zu ~17.000 mit Bulk-RNA-Seq, alles zu ~1% der Kosten.
📈Schlüsselergebnisse
- Mechanismusbasierte Clusterbildung:
t-SNE-Analyse von DRUG-seq-Daten gruppierte Verbindungen nach gemeinsamen Wirkmechanismen (MoA) und unterschied sogar zwischen Inhibitoren, die dasselbe Protein anvisieren. - Dosis-Wirkungs-Sensitivität:
Die Veränderungen der Genexpression waren eindeutig dosisabhängig und entsprachen dem erwarteten Verhalten der Verbindungen über mehrere Zielgruppen hinweg. - CRISPR vs. Verbindungsvergleich:
Nachgewiesen, dass DRUG-seq die transkriptionalen Ergebnisse von CRISPR-vermitteltem Gen-Knockout im Vergleich zur pharmakologischen Hemmung desselben Ziels unterscheiden kann.
🧠Fazit & Auswirkungen
- Skalierbarkeit und Kosten-effizienz:
Hochdurchsatz-Profilierung über Hunderte von Verbindungen bei minimalen Kosten pro Probe (~2–4 $). - Reiche mechanistische Einsicht:
Robuste MoA-Clustering validiert transkriptionale Signaturen, ermöglicht die Erkennung von Off-Target-Effekten und die Validierung von Zielen. - Flexible Assay-Kompatibilität:
Nachgewiesene Anwendbarkeit sowohl in chemischen als auch in genetischen Störkontexten, wodurch DRUG-seq ein vielseitiges Werkzeug für Arzneimittelentdeckungspipelines ist.
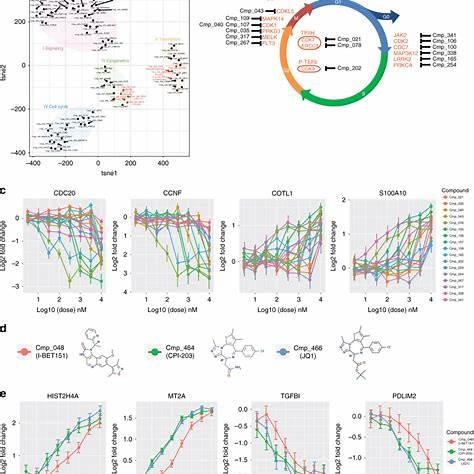 Abbildung: Mechanistische Clusterbildung von DRUG-seq
Abbildung: Mechanistische Clusterbildung von DRUG-seq
t-SNE-Diagramm, das die Clusterbildung von mit Verbindungen behandelten Proben zeigt. Jeder Punkt repräsentiert eine Kombination aus Verbindung und Dosis, farblich codiert nach Wirkmechanismus (z. B. CDK-Inhibitoren, epigenetische Modulatoren). Die Cluster spiegeln funktionale Gruppen wider (Abb. 3a in Ye et al. 2018).


