
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Whole-Genome-Bisulfid-Sequenzierung (WGBS)
Als führender Anbieter von NGS-Dienstleistungen bietet CD Genomics ein integriertes Portfolio von MethylierungssequenzierungsdiensteDie gesamte Genom-Bisulfid-Sequenzierung (WGBS) ist eine effektive und zuverlässige Strategie, um individuell methyliertes Cytosin auf genomweiter Ebene zu identifizieren. Mit über 10 Jahren Erfahrung und der neuesten Technologie Next-Generation-Sequenzierung Plattformen, wir können Ihre Projektanforderungen und Budgets bei der Erforschung des Methyloms vollständig erfüllen.
Was ist die Whole-Genome-Bisulfid-Sequenzierung?
In der Säugetierbiologie beinhaltet 5-Methylcytosin eine kovalente Bindung zwischen einer Methylgruppe und der 5'-Kohlenstoffposition von Cytosin, wodurch zusätzliche kapazitive Eigenschaften für die Signaltransduktion und regulatorische Funktionen verliehen werden. Als eine der vorherrschenden epigenetischen Modifikationen spielt 5-Methylcytosin eine entscheidende Rolle in zahlreichen biologischen Prozessen, einschließlich der Genstilllegung, der Repression von Transposons, der Hemmung repetitiver Sequenzen, der genomischen Prägung und der Inaktivierung des X-Chromosoms. Die Erkennung und Quantifizierung von Methylierung sind grundlegend wichtig für das Verständnis der Genexpression und anderer Prozesse, die der epigenetischen Regulation unterliegen.
DNA-Methylierung stellt einen wesentlichen Bestandteil von Epigenetik, spielt eine wesentliche Rolle bei der Aufrechterhaltung normaler Zellfunktionen, genetischer Prägung, embryonaler Entwicklung und dem Ausbruch der menschlichen Tumorigenese. WGBS, das eine Bisulfit-Konversion von unmodifizierten Cytosinen zu Uracilen in genomischer DNA verwendet, ermöglicht die Erstellung eines umfassenden Methylierungsprofils. Durch die Durchführung einer Hochdurchsatz-Nachsequenzierung der konvertierten DNA und den Vergleich mit einem Referenzgenom ermöglicht die WGBS-Methode eine genomweite Analyse von Methylierungsmetriken mit einer Auflösung auf Einzelbasenebene und bietet hohe Präzision. Ihre breitere Anwendung reicht von der Untersuchung der grundlegenden Mechanismen der Zell-Differenzierung und Gewebeentwicklung bis hin zur Förderung von Bereichen wie landwirtschaftlicher Züchtung, menschlicher Gesundheit und Krankheitsbehandlung.
WGBS ist die bevorzugte Methode zur Erstellung umfassender Karten der DNA-Methylierung auf der Ebene einzelner Basen. Dieser Ansatz, der oft als der "Goldstandard" für Methylierungssequenzierung bezeichnet wird, zeichnet sich durch seine hohe Durchsatzrate, Zeit-effizienz, Einzelbasisauflösung und umfassende Abdeckung aus.
Prinzip von WGBS
Das Prinzip der WGBS beruht auf der Behandlung mit schwerem Bisulfat, um unmethylierte Cytosine im Genom in Uracile umzuwandeln, die nach der PCR-Amplifikation zu Thyminen werden und somit von methylisierten Cytosinen unterschieden werden können. In Kombination mit Hochdurchsatz-Sequenzierungstechnologie und Referenzsequenzvergleichen kann die Methylierung an CpG/CHG/CHH-Stellen bestimmt werden, was es besonders geeignet macht, Methylierungskarten des gesamten Genoms mit einer Auflösung auf Einzelbasenebene zu erstellen. Für weitere detaillierte Informationen siehe den Artikel, "Prinzipien und Arbeitsablauf der Whole Genome Bisulfite-Sequenzierung.
Vorteile von WGBS
- Geeignet für die Forschung an Menschen sowie den meisten Tieren und Pflanzen (mit bekannten Referenzgenomen).
- Hochintegrierte DNA-Methylierungsmuster mit Einzelbasenauflösung.
- Maximal umfassende Informationen zur gesamten Genom-Methylierung erhalten und Methylierungsmuster genau kartieren.
- Bereitstellung von Einblicken in das Schicksalsengagement und die Reprogrammierung von Genzellen sowie in die Genregulation.
- Identifizierung neuer epigenetischer Marker und Krankheitsziele.
Anwendung von WGBS
- Regulation der Genexpression
- Epigenetische Heterogenität
- Umwelt und Epigenetik
- Embryonale Entwicklung
- Krankheitsforschung
- Genetische Abdrücke
- Tumorforschung
- Forschung zur Genomstabilität
- Evolutionsbiologische Forschung
Verschiedene Techniken zur Erkennung von DNA-Methylierung
450K/850K
Diese Technologie scannt und erkennt etwa 450.000 und 850.000 wichtige CpG-Stellen, die im Genom durch Chip-Scanning bekannt sind. Sie weist eine hohe Genauigkeit und Wiederholbarkeit auf, ist jedoch auf menschliche Proben beschränkt und kann neue CpG-Stellen außerhalb des Chips im Genom nicht erkennen.
MeDIP-seq
Methyliertes DNA-Immunpräzipitations-Sequenzieren (MeDIP-seq) Bereichert und sequenziert Regionen, die einer Methylierung unterliegen, und ermittelt den Methylierungsstatus der am stärksten methylisierten Regionen im gesamten Genom. Es fehlt an Einzelbasenauflösung und absoluter Quantifizierung der Methylierungsniveaus und bietet lediglich relative Methylierungsniveaus. Es eignet sich zur Erforschung des genomweiten Methylierungsstatus, ohne dass eine Einzelbasenauflösung erforderlich ist.
WGBS
Diese Technik kann effektive CpG-Stellen erkennen, die über 75 % aller CpG-Stellen im gesamten Genom erreichen, und erzielt eine Einzelbasisauflösung. Sie eignet sich für die systematische und umfassende Untersuchung des genomweiten Methylierungsstatus.
RRBS
Reduzierte Repräsentation Bisulfit-Sequenzierung (RRBS) Erfasst den Methylierungsstatus von Regionen, die reich an CG-Typ-Stellen sind (hauptsächlich regulatorische Regionen) im Genom, mit Einzelbasenauflösung und hoher Kosteneffizienz. Es eignet sich zur Untersuchung des Methylierungsstatus von regulatorischen Regionen.
Ziel-BS
Zielgerichtete Bisulfid-Sequenzierung Entwirft Primer für Zielfragmente, führt die PCR-Amplifikation nach der BS-Konversion durch und erstellt schließlich Bibliotheken für die Next-Generation-Sequenzierung. Die Sequenzierungstiefe der Zielregionssequenzierung kann von Hunderten bis zu Zehntausenden reichen, was eine präzise Erkennung der Methylierungsniveaus an C-Stellen innerhalb der Zielregionen ermöglicht. Es wird für die anschließende Methylierungsverifizierung von Zielgenen verwendet.
Der Illumina 935K DNA-Methylierungsassay-Service ist ein ausgeklügeltes Werkzeug, das für die Erforschung der DNA-Methylierung entwickelt wurde. Der Infinium MethylationEPIC v2.0 BeadChip (935K) ermöglicht eine Hochdurchsatzanalyse von etwa 935.000 CpG-Standorten, die im gesamten menschlichen Genom verteilt sind. Diese aktualisierte Version bietet eine verbesserte Abdeckung und Präzision und gewährleistet die Übereinstimmung mit den neuesten genomischen Datenbanken. Zu den bemerkenswerten Eigenschaften dieses Geräts gehören die Möglichkeiten zur Ganzgenomanalyse, bestätigte Zuverlässigkeit und breite Kompatibilität mit verschiedenen Probenkategorien, einschließlich formalinfixierter, paraffin-eingebetteter Gewebe. Der Service richtet sich speziell an umfassende epigenetische Studien und bietet Forschern die Mittel, um zugrunde liegende Krankheitsmechanismen zu entschlüsseln und potenzielle Biomarker zu identifizieren.
Die Illumina Methylierungs-Screening-Array 270K (MSA270K) stellt eine hochmoderne Innovation dar, die speziell für sorgfältige Methylierungsuntersuchungen innerhalb bestimmter Krankheitskohorten und umfassender Gesundheitsbewertungen entwickelt wurde. Mit etwa 270.000 Methylierungsstellen bietet dieses Werkzeug eine verbesserte Effizienz, indem es einen Durchsatz von 48 Proben pro Array ermöglicht, was im Vergleich zu seinem Vorgänger eine Ver sechsfachung darstellt. Es befähigt Wissenschaftler, Fortschritte in explorativen Bestrebungen im Zusammenhang mit krankheitsphänotypischen Mustern, den Umwelteinflüssen auf die Epigenetik und Veränderungen in der Methylierung, die mit dem Altern verbunden sind, zu erzielen. Der MSA270K stärkt unser Verständnis von DNA-Methylierung und deren Auswirkungen auf die menschliche Gesundheit und Krankheitszustände erheblich. Dies katalysiert Paradigmenwechsel im grundlegenden Verständnis unseres angeborenen genetischen Bauplans und eröffnet potenzielle zukünftige therapeutische Wege.
WGBS-Workflow
Unser hochqualifiziertes Expertenteam und die strenge Qualitätskontrolle nach jedem Verfahren gewährleisten umfassende und präzise Ergebnisse. Vor der Hochdurchsatz-Sequenzierung wird die DNA-Probe mit Cytosin-Bisulfid-Umwandlung bearbeitet, gefolgt von der Markierung an den 5'- und 3'-Enden sowie der Einführung von Illumina-Adaptern durch PCR-Amplifikation.
Um die Genauigkeit und Zuverlässigkeit von Sequenzierungsdaten sicherzustellen, führt CD Genomics jeden experimentellen Schritt von der DNA-Probenentnahme bis zum Erhalt des endgültigen Datenberichts sorgfältig durch strenge Qualitätskontrollen. Dieser umfassende Ansatz gewährleistet grundsätzlich eine hohe Datenqualität. Die Sicherstellung hochwertiger Daten ist die grundlegende Voraussetzung für genaue, umfassende und glaubwürdige Ergebnisse. Bioinformatik Analyse.
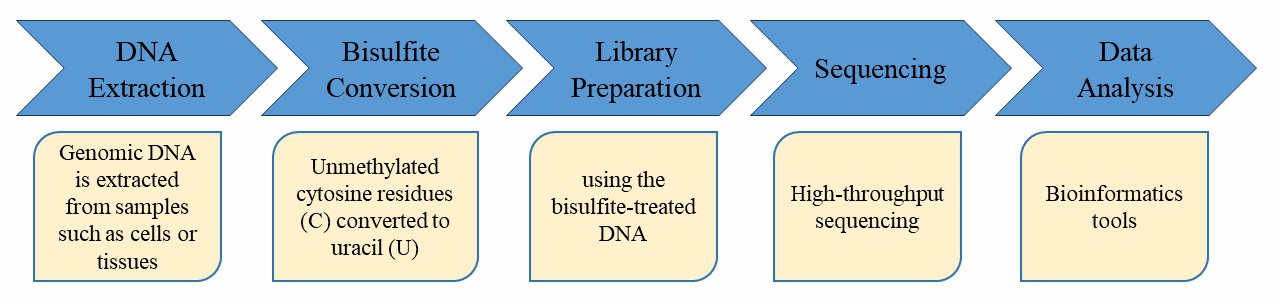
Dienstspezifikation
Musteranforderungen und Vorbereitung
|
|
 |
Sequenzierung
|
 |
Datenanalyse Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an:
|
Analyse-Pipeline
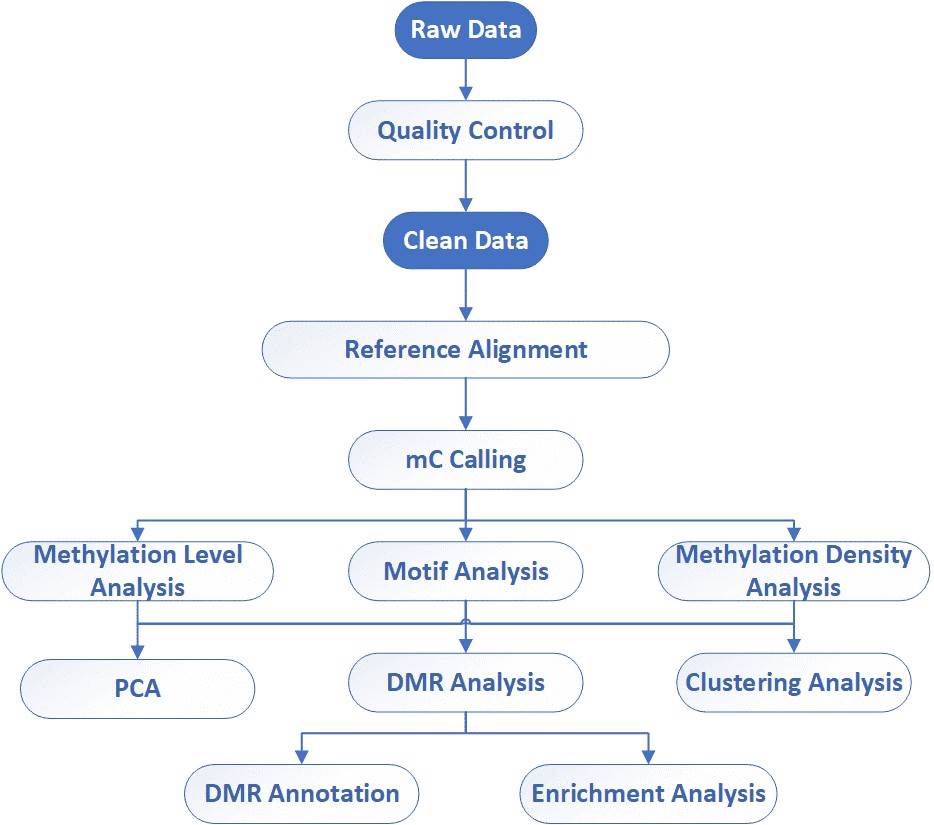
Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details in WGBS für Ihr Schreiben (Anpassung)
Durch den Einsatz modernster Next-Generation-Sequenzierungsplattformen und unsere umfangreiche Erfahrung garantieren wir Ihnen hochwertige Daten und integrierte bioinformatische Analysen. Wenn Sie daran interessiert sind, was CD Genomics mit der gesamten Genom-Bisulfid-Sequenzierung tun kann, zögern Sie bitte nicht, uns zu kontaktieren. Wir freuen uns darauf, Ihr Projekt voranzubringen.
Referenzen:
- Huang, K., & Fan, G. DNA-Methylierung bei der Zell-Differenzierung und Reprogrammierung: eine aufkommende systematische Sicht. Regenerative Medizin, 2010, 5(4), 531-544.
- Beck D, Ben Maamar M, Skinner M K. Vergleich der Methoden zur Analyse der genomweiten CpG-Dichte und DNA-Methylierung (MeDIP, RRBS und WGBS). Epigenetik, 2022, 17(5): 518-530.
- Abante J, Fang Y, Feinberg A P, et al. Nachweis von haplotypabhängiger allelspezifischer DNA-Methylierung in WGBS-Daten. Naturkommunikationen, 2020, 11(1): 5238.
- Suzuki M, Liao W, Wos F, et al. Ganzgenom-Bisulfid-Sequenzierung mit verbesserter Genauigkeit und Kosten. Genomforschung, 2018, 28(9): 1364-1371.
Demonstrationsergebnisse
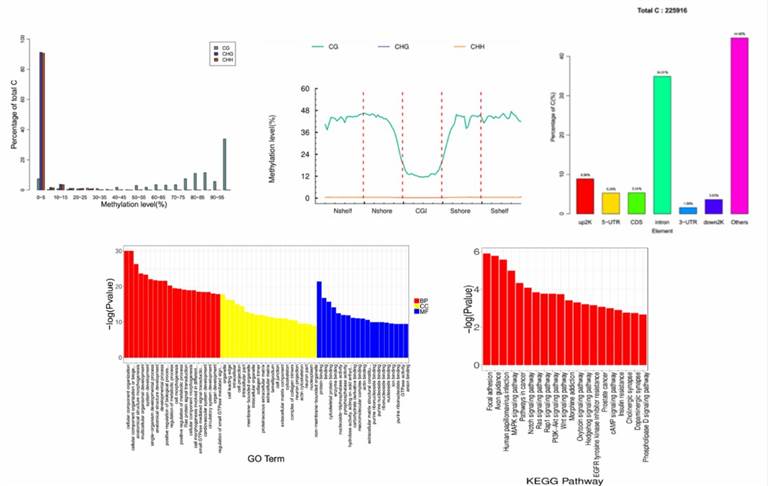
Häufig gestellte Fragen zur Whole Genome Bisulfite-Sequenzierung
1. Welche Arten eignen sich für die gesamte Genom-Bisulfid-Sequenzierung?
Die Arten, die einer Genom-Bisulfid-Sequenzierung unterzogen werden, sollten die folgenden drei Bedingungen erfüllen:
a. Eukaryoten.
b. Sein Referenzgenom wurde mindestens auf das Scaffold-Niveau assembliert.
c. Relativ vollständige Genomannotationen.
2. Was sind die Hauptinhalte der fortgeschrittenen bioinformatischen Analyse in WGBS?
Die Analyse der Korrelation zwischen Methylierungslevels und Transkriptionslevels stellt einen wesentlichen Bestandteil fortgeschrittener bioinformatischer Analysen in der gesamten Genom-Methylierungssequenzierung dar. Sie umfasst hauptsächlich vier Aspekte:
- Methylierungsniveaus von differentiell exprimierten Genen.
- Methylierungsniveaus von unterschiedlich exprimierten transponierbaren Elementen.
- Beziehung zwischen Methylierungsmodifikationen und Expressionsregulation.
- Expressionsanalyse von methylierten und nicht methylierten Genen.
3. Was sind die Vorteile von WGBS im Vergleich zu anderen Methyliierungs-Sequenzierungsmethoden?
Im Bereich der DNA-Methylierungsequenzierung umfassen die aktuellen Hauptmethoden die Whole Genome Bisulfite Sequenzierung (WGBS), Methylierte DNA-Immunpräzipitation (MeDIP)und Reduzierte Repräsentation Bisulfid-Sequenzierung (RRBS)WGBS bietet den Vorteil, den Methylierungsstatus an nahezu jeder CpG-Stelle bewerten zu können. Diese Bewertung kann sich auf Bereiche mit niedriger CpG-Dichte erstrecken, einschließlich intergenerischer "Genwüsten", teilweise methylierten Domänen und entfernten regulatorischen Elementen. Darüber hinaus kann WGBS absolute DNA-Methylierungsniveaus und den Methylierungssequenzkontext aufdecken. Es sollte jedoch beachtet werden, dass MeDIP keine Einzelbasenauflösung der Methylierung erreichen kann und RRBS typischerweise Bereiche mit einem höheren Grad an Methylierung für die Analyse auswählt, was es ausschließt, den Methylierungsstatus auf der Ebene des gesamten Gens zu erhalten.
4. Was sind die Einschränkungen von WGBS?
- Dieses Verfahren kann nur 5mC oder 4mC nachweisen und ist nicht in der Lage, 6mA zu detektieren.
- Die Behandlung mit Bisulfit führt zu einer erheblichen Fragmentierung der DNA, die die Amplifikation beeinträchtigen kann.
- Die Umwandlung von unmethylierter Cytosine in Uracile während der Bisulfitbehandlung erhöht die Komplexität der Bibliotheksvorbereitung. Unvollständige Umwandlung kann ebenfalls Verzerrungen in die Ergebnisse einführen.
- Die Methode kann 5mC und 5hmC nicht effektiv unterscheiden.
Referenzen:
- Beck D, Ben Maamar M, Skinner M K. Vergleich von genomweiten CpG-Dichten und Methoden zur Analyse der DNA-Methylierung (MeDIP, RRBS und WGBS). Epigenetik, 2022, 17(5): 518-530.
- Suzuki M, Liao W, Wos F, et al. Ganzgenom-Bisulfid-Sequenzierung mit verbesserter Genauigkeit und Kosten. Genomforschung, 2018, 28(9): 1364-1371.
Fallstudien zur Whole Genome Bisulfite-Sequenzierung
Abweichende DNA-Methylierungsmuster im Zusammenhang mit der Genexpression bei Reisvarianten mit unterschiedlichen Reaktionen auf Trockenstress und Salinitätsstress
Journal: Wissenschaftliche Berichte
Impactfaktor: 4,259
Veröffentlicht: 09. Oktober 2015
Hintergrund
DNA-Methylierung spielt eine zentrale Rolle bei der Regulation der Genaktivität und der Chromatinstruktur als Reaktion auf Umweltbedingungen. Das Verständnis der genomweiten DNA-Methylierung liefert Einblicke in die regulatorischen Mechanismen der Reaktion/Anpassung an abiotischen Stress.
Materialien: Drei Reis (Oryza sativa) Sorten, IR64 (empfindlich gegenüber Trockenheit und Salinität), Pokkali (salzresistent) und Nagina 22 (trockenheitstolerant).
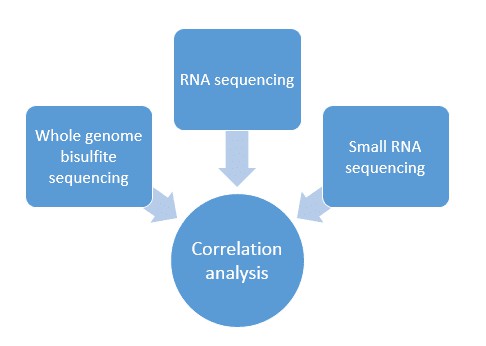
Methoden
- Nanodrop-Spektrophotometer
- Qubit-Fluorimeter
- Whole-Genome-Bisulfid-Sequenzierung:
- PCR-Amplifikation
- Bibliotheksqualitätsanalyse
- Primer Express (v3.0)
- Echtzeit-PCR-System
- Gene-Ontologie (GO) Anreicherungsanalyse
- Methylierungs-Expressions-Korrelationsanalyse
Ergebnisse
Forscher untersuchten die Unterschiede zwischen den drei Sorten in den DNA-Methylierungsmustern, DMRs (differenziell methylierten Regionen) und der Genexpression und erkundeten weiter die zugrunde liegenden Zusammenhänge.
Differenzielle Methylierung ist mit differenzieller Genexpression in verschiedenen Sorten gekoppelt.
Im Vergleich zu allen Genen zeigen die Gene, die proximal zu hoch methylierten differentiell methylierten Regionen (DMRs) liegen, ein niedrigeres Niveau der Transkriptabundanz, was auf eine Herunterregulierung hindeutet. Im Gegensatz dazu weisen Gene in der Nähe von niedrig methylierten DMRs ähnliche oder leicht höhere Niveaus der Transkriptabundanz auf, was auf eine Hochregulierung hindeutet. Bei DMRs scheint die DNA-Methylierung auch mit dem Ein/Aus-Status der Genexpression korrelieren, wie durch 14,5 % in N22/IR64 und 7,2 % in PK/IR64 belegt wird.
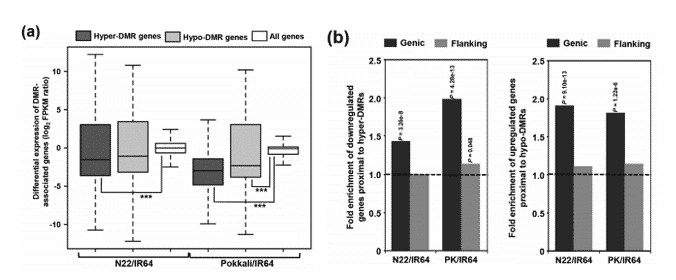 Abbildung 1. Beziehung zwischen differentieller Methylierung und Transkriptionshäufigkeit von protein-codierenden Genen.
Abbildung 1. Beziehung zwischen differentieller Methylierung und Transkriptionshäufigkeit von protein-codierenden Genen.
2. Differenzielle Methylierung von Genen, die mit der Stressreaktion und der epigenetischen Regulation der Genexpression assoziiert sind.
Der differentielle Methylierungsgrad und die entsprechenden differentiellen Genexpressionen von Reisgenen in N22/IR64 und PK/IR64 sind in Abbildung (a) unten dargestellt. Die GO-Analyse der mit DMR assoziierten Gene in N22/IR64 und PK/IR64 ergab eine signifikante Anreicherung von Genen, die an der Stressreaktion beteiligt sind.
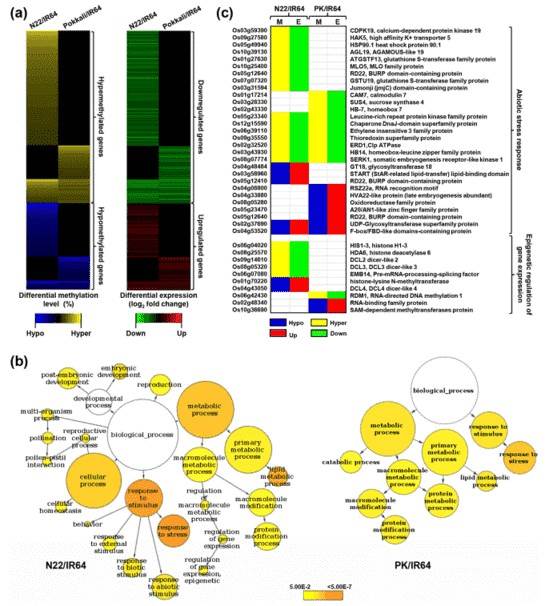 Abbildung 2. Differenzielle Methylierung, Transkriptmenge und GO-Anreicherung von proteincodierenden Genen.
Abbildung 2. Differenzielle Methylierung, Transkriptmenge und GO-Anreicherung von proteincodierenden Genen.
Der Methylierungsstatus von TEs steht in Zusammenhang mit der Transkription proximaler protein-codierender Gene.
Die Anreicherung von TEs war erheblich höher für DMR-assoziierte DEGs im Vergleich zu non-DEGs sowohl für N22/IR64 (P-Wert 1,49e-05) als auch für PK/IR64 (P-Wert 5,26e-05) (a). Signifikant höhere Unterschiede im Methylierungsniveau von DMRs sind mit TEs im Vergleich zu protein-codierenden Genen für PK/IR64 assoziiert (b).
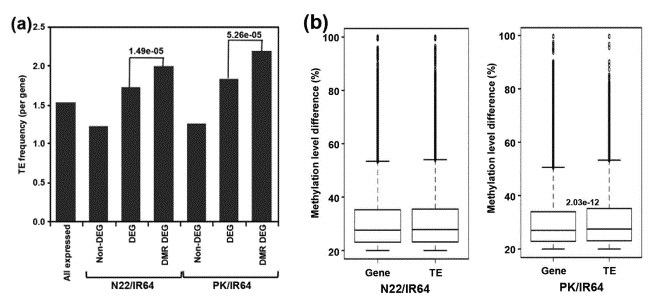 Abbildung 3. Korrelation zwischen differentieller Expression und Häufigkeit/Methylierungsgrad von TEs.
Abbildung 3. Korrelation zwischen differentieller Expression und Häufigkeit/Methylierungsgrad von TEs.
Fazit
Die Ergebnisse deuten auf die potenzielle Rolle von kultivarspezifischen DNA-Methylierungsmustern als entscheidenden Regulationsmechanismus für das Erkennen und Reagieren auf Stressbedingungen durch Modulation der Expression stressreaktiver Gene hin. Die Untersuchungen legen nahe, dass die Variabilität in der Dürre- oder Salztoleranz im Reisgenpool von dem Ausmaß und den Mustern der DNA-Methylierung abhängt. Und die Beweise deuten darauf hin, dass die DNA-Methylierung eine wichtige Rolle in der Reaktion auf abiotischen Stress spielt, indem sie die Expression einer Reihe von stressreaktiven Genen im Reis hauptsächlich durch Methylierung oder Demethylierung von proximalen Transposons reguliert.
Referenz:
- Garg R, Chevala V V S N, Shankar R u. a. Abweichende DNA-Methylierungsmuster in Verbindung mit der Genexpression bei Reisvarianten mit unterschiedlichen Reaktionen auf Trockenheits- und Salztstress. Wissenschaftliche Berichte, 2015, 5.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Methylierung im CHH-Kontext ermöglicht die Vorhersage von Rekombination in Reis.
Zeitschrift: Internationale Zeitschrift für Molekulare Wissenschaften
Jahr: 2022
Integration von DNA-Methylierungsdaten des gesamten Genoms mit RNA-Sequenzierungsdaten zur Identifizierung von Markern für die Fruchtbarkeit von Bullen
Zeitschrift: Tiergenetik
Jahr: 2020
Fettreiche Diäten während der Schwangerschaft führen zu Veränderungen der DNA-Methylierung und Proteinexpression im Pankreasgewebe der Nachkommen: Ein Multi-Omics-Ansatz
Internationale Zeitschrift für Molekulare Wissenschaften
Jahr: 2024
KMT2A assoziiert mit dem PHF5A-PHF14-HMG20A-RAI1 Subkomplex in Stammzellen des Bauchspeicheldrüsenkrebses und reguliert epigenetisch deren Eigenschaften.
Zeitschrift: Nature Communications
Jahr: 2023
Cholestensäure als endogener epigenetischer Regulator verringert die Lipidansammlung in Hepatozyten in vitro und in vivo.
Journal: Amerikanisches Journal für Physiologie - Gastrointestinale und Leberphysiologie
Jahr: 2024
DNA-Methyltransferase, die an der Wiederherstellung der Konidienbildung durch aufeinanderfolgende Pflanzenpassagen bei phänotypisch degeneriertem Metarhizium beteiligt ist.
Zeitschrift: Angewandte Mikrobiologie und Biotechnologie
Jahr: 2020
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


