
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Hi-SSRseq
Die Einführung von Hi-SSRseq
Mikrosatelliten (kurze tandemwiederholungen, STR oder einfache Sequenzwiederholungen, SSR) sind weit verbreitete Marker in der Populationsgenetik. Obwohl eine genaue und effiziente Genotypisierung von SSRs die Grundlage für SSRs als effektiven genetischen Marker mit verschiedenen Anwendungen bildet, hat das Fehlen einer Hochdurchsatztechnologie für die SSR-Genotypisierung ihre Verwendung als genetische Ziele in vielen Kulturpflanzen eingeschränkt. Einzelnukleotidpolymorphismen (SNPs) oder Insertionen/Deletion (Indel)-Polymorphismen in der Nukleotidsequenz dieses Fragments, entweder innerhalb des repetitiven Arrays oder in den flankierenden Regionen (FR), bleiben allein durch Längenmessung unentdeckt. Darüber hinaus könnten Indels in den flankierenden Regionen fälschlicherweise mit Größenmutationen der SSR verwechselt werden.
Als Folge kann die traditionelle Bewertung der Fragmentlängen zu einer Unterschätzung der genetischen Variabilität, ungenauen Ergebnissen oder sogar falschen evolutionären Interpretationen führen. Um solche Fehler zu vermeiden, sind Informationen über die Nukleotidsequenz jedes Allels erforderlich. CD Genomics stellte eine Technologie namens Hi-SSRseq zur Verfügung, die die multiplexe Amplifikation traditioneller SSRs mit Hochdurchsatz-SequenzierungDieses Verfahren kann zahlreiche SSR-Loci in Hunderten von Proben mit hochgenauen Ergebnissen genotypisieren, dank der umfangreichen Abdeckung durch Hochdurchsatz-Sequenzierung, die auch die Kosten und die Zeit für das Genotyping erheblich reduziert, und der Vergleich zwischen den Proben kann direkt auf der Basensequenz basieren.
Unsere Ziele waren (a) die Generierung von Nukleotidsequenzdaten mehrerer Nicht-Modell-Pflanzenarten, für die zuvor keine genomischen Daten existierten, sowohl aus den SSR- als auch aus den flankierenden Regionen, (b) die Erfassung der Länge des repetitiven Bereichs sowie der SNP- und Indel-Variationen innerhalb des SSR und der FR, (3) die Schätzung des Ausmaßes der molekular zugänglichen Größenhomoplasie jedes Locus und (4) der Vergleich des Grades der genetischen Variabilität zwischen verschiedenen Datensätzen basierend auf der Anzahl der Wiederholungseinheiten, der Fragmentlänge und der Sequenzidentität.
Anwendungen von Hi-SSRseq
- Genetische Analyse
- Feinabstimmung
- Quantitative Trait Locus (QTL) Mapping
- Marker-gestützte Selektion (MAS) Züchtung
Hauptmerkmale und Vorteile von Hi-SSRseq
- Hoher Durchsatz: Viele SSR-Loci können in Hunderten von Proben mit hochgenauen Ergebnissen genotypisiert werden.
- Kostenwirksam: Die Kosten für den Test sind deutlich geringer als die der meisten anderen. SSR-Genotypisierung Plattformen.
- Vereinfachter Handlungsablauf.
- Genauere Ergebnisse: Vermeidung von Unterschätzungen der genetischen Variabilität, falschen evolutionären Interpretationen.
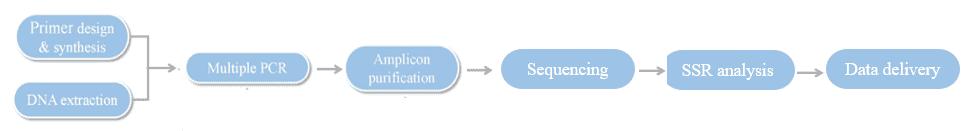
Hi-SSRseq Arbeitsablauf
Dienstspezifikationen
Musteranforderungen
|
|
 Klicken |
Sequenzierungsstrategie
|
 |
Bioinformatische Analyse Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an:
|
Analyse-Pipeline

Liefergegenstände
- Rohdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details in Hi-SSRseq für Ihr Schreiben (Anpassung)
Mit modernsten Sequenzierungsplattformen und enger Zusammenarbeit mit hochqualifizierten Technikern und Wissenschaftlern in den verschiedenen Abteilungen von CD Genomics bieten wir eine Hi-SSRseq-Technik an, die es ermöglicht, Hunderte von Individuen gleichzeitig an vielen maßgeschneiderten SSR-Loci zu genotypisieren, indem Multiplex-PCR und Illumina-Sequenzierung kombiniert werden. Wenn Sie zusätzliche Anforderungen oder Fragen haben, zögern Sie bitte nicht, uns zu kontaktieren.
Referenzen
- Petra Šarhanová, Simon Pfanzelt, Ronny Brandt, Axel Himmelbach und Frank R. Blattner. SSR-seq: Genotypisierung von Mikrosatelliten mittels Next-Generation-Sequencing zeigt ein höheres Maß an Polymorphismus im Vergleich zur traditionellen Fragmentgrößenbestimmung. Ökologie und Evolution. 2018;8:10817–10833.
- Jingjing Yang, Jian Zhang, Ruixi Han, Feng Zhang, Aijun Mao, Jiang Luo, Bobo Dong, Hui Liu, Hao Tang, Jianan Zhang und Changlong Wen. Ziel-SSR-Seq: Eine neuartige SSR-Genotypisierungstechnologie, die mit perfekten SSRs in der genetischen Analyse von Gurkensorten verbunden ist. Frontiers in Plant Science. 2019; 10:1-12.
Demonstrationsergebnisse
Teilweise Ergebnisse sind unten aufgeführt:

Hi-SSRseq häufig gestellte Fragen (FAQs)
1. Welche Art von Unterstützung ist während des Hi-SSRseq-Prozesses verfügbar?
Wir bieten umfassende Unterstützung während des gesamten Hi-SSRseq-Prozesses, einschließlich:
- Technische Unterstützung: Anleitung zur Probenvorbereitung, Bibliothekskonstruktion und Dateninterpretation.
- Datenanalyse: Expertenanalyse von Sequenzierungsdaten und Identifizierung von SSRs.
- Beratung: Regelmäßige Updates und Konsultationen, um Fragen oder Probleme zu klären.
2. Wie unterscheidet sich Hi-SSRseq von der traditionellen SSR-Genotypisierung?
Traditionell SSR-Genotypisierung oft auf die Bewertung der Fragmentlängen angewiesen, die durch Probleme wie Größenhomoplasie und die Unfähigkeit, Einzel-Nukleotid-Polymorphismen (SNPs) oder Insertionen/Löschungen (Indels) in angrenzenden Regionen zu erkennen, eingeschränkt sein kann. Hi-SSRseq hingegen verwendet Hochdurchsatz-Sequenzierung um detaillierte Nukleotidsequenzdaten für jedes SSR-Lokus bereitzustellen, die Genauigkeit zu verbessern und häufige Fehlinterpretationen, die sich nur auf die Fragmentlänge beziehen, zu vermeiden.
3. Wie verbessert Hi-SSRseq die genetische Analyse im Vergleich zu traditionellen Methoden?
Hi-SSRseq bietet detaillierte Sequenzdaten, die die genetische Analyse verbessern durch:
- Größenhomoplasie reduzieren: Missverständnisse im Zusammenhang mit der Fragmentlänge ansprechen.
- Erkennung von SNPs und Indels: Identifizierung von Variationen in SSR und flankierenden Regionen.
- Genauigkeit erhöhen: Präzise evolutionäre Interpretationen und Schätzungen der genetischen Variabilität anbieten.
4. Wie werden SSRs in Hi-SSRseq identifiziert und analysiert?
SSRs werden identifiziert, indem die Sequenzierungsdaten analysiert werden, um sich wiederholende DNA-Sequenzen zu erkennen. Bioinformatik-Tools und -Algorithmen werden verwendet, um:
- Sequenzen ausrichten: Ordnen Sie die Reads einem Referenzgenom zu.
- Wiederholungen erkennen: SSRs basierend auf Wiederholungseinheiten und Länge identifizieren und klassifizieren.
- Daten filtern: Wählen Sie signifikante SSRs basierend auf vordefinierten Kriterien aus.
- Annotieren: Bereitstellung funktionaler und genomischer Kontexte für jedes SSR.
Hi-SSRseq Fallstudien
Das genetische Erbe von Fragmentierung und Übernutzung im bedrohten medizinischen afrikanischen Pfefferbaum, Warburgia salutaris
Journal: Wissenschaftliche Berichte
Impactfaktor: 4,997
Veröffentlicht: 12. November 2020
Hintergrund
Heilpflanzen sind weltweit von entscheidender Bedeutung, insbesondere in Entwicklungsländern. Die reiche Pflanzenvielfalt in Subsahara-Afrika ist durch menschliche Aktivitäten bedroht. Warburgia salutaris, eine wichtige Heilpflanze im südlichen Afrika, ist aufgrund von Überernte akut gefährdet. Forscher entwickelten SSR-Marker, um die genetische Vielfalt und Struktur der Pflanze zu bewerten, wobei der Fokus auf Mosambik lag, um Maßnahmen zum Schutz und zur Wiedereinführung zu informieren.
Materialien & Methoden
Probenvorbereitung
- Immergrüner Baum
- Warburgia salutaris
- Junge unbeschädigte Blätter
- DNA-Extraktion
Methode
- SSR-Entwicklung
- Hi-SSRseq
- Illumina Hiseq 2500
- Bestimmung der Allelgröße
- Schätzungen der genetischen Vielfalt
- Populationsgenetische Struktur und Differenzierung
Ergebnisse
1. Genetische Vielfalt
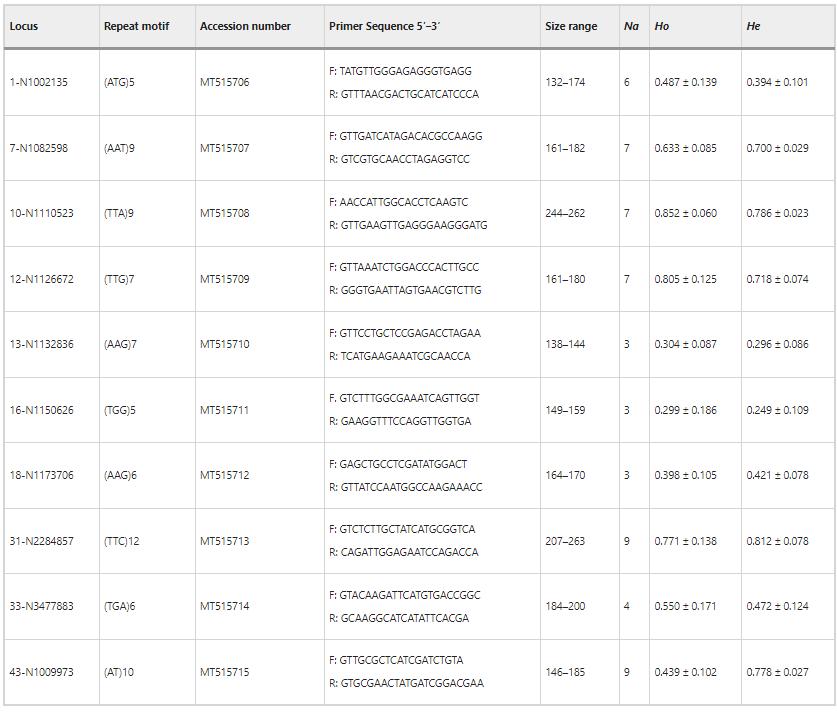
Die Studie identifizierte 58 Allele an 10 SSR-Loci in Warburgia salutaris, mit Allelzahlen, die von drei bis neun pro Locus reichten. Die durchschnittliche beobachtete Heterozygosität variierte von 0,299 bis 0,852, während die erwartete Heterozygosität von 0,249 bis 0,812 reichte. Die Diversität war im LM-Gebiet am höchsten, mit einem höheren Shannon-Diversitätsindex im Vergleich zu den TR- und FC-Gebieten. Der polymorphe Informationsgehalt war hoch, und die Inzuchtkoeffizienten waren in allen Gebieten niedrig.
Tabelle 1 Eigenschaften und genetische Diversitätsstatistiken der 10 polymorphen Mikrosatellitenmarker, die für entwickelt wurden Warburgia salutaris.
2. Populationsgenetische Struktur und Differenzierung
Die STRUCTURE-Analyse ergab zwei Hauptgenetische Cluster: eines in den LM- und TR-Gebieten und ein anderes im FC-Gebiet. PCoA- und Nachbarverbindungsbaum-Analysen unterstützten diese Ergebnisse und zeigten, dass die Populationen aus FC deutlich getrennt waren, während die aus LM und TR stärker vermischt waren. Die paarweisen FST-Werte wiesen auf eine moderate genetische Differenzierung zwischen FC und den anderen Gebieten hin, mit geringerer Differenzierung zwischen TR und LM.
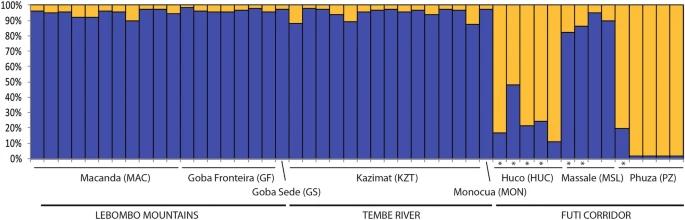 Abb. 1. Populationsstruktur von Warburgia salutaris basierend auf 10 SSRs und unter Verwendung des besten Zuordnungsergebnisses, das von STRUCTURE abgerufen wurde.
Abb. 1. Populationsstruktur von Warburgia salutaris basierend auf 10 SSRs und unter Verwendung des besten Zuordnungsergebnisses, das von STRUCTURE abgerufen wurde.
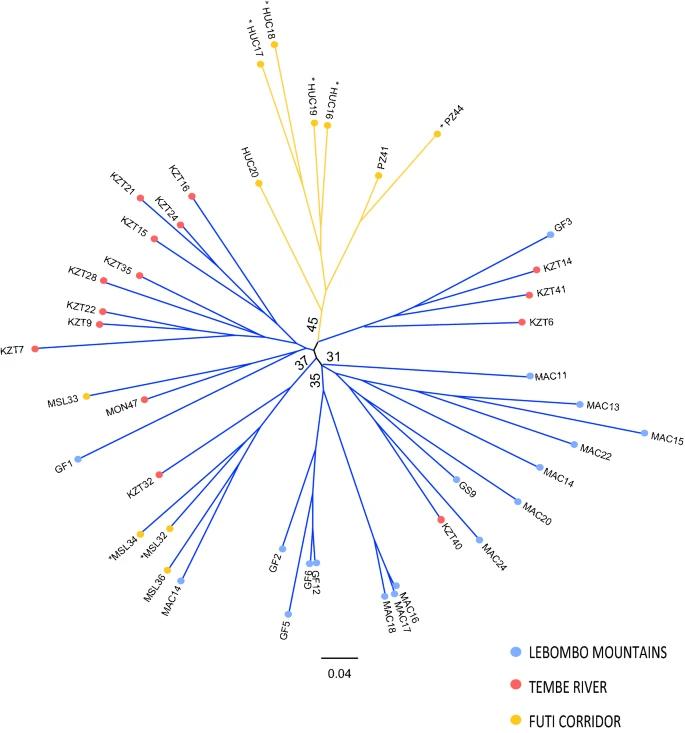 Abb. 2. Unverzweigter Nachbarverbindungsbaum der untersuchten Warburgia salutaris basierend auf Neis genetischer Distanz.
Abb. 2. Unverzweigter Nachbarverbindungsbaum der untersuchten Warburgia salutaris basierend auf Neis genetischer Distanz.
Fazit
Warburgia salutaris zeigt eine hohe genetische Vielfalt und Vermischung trotz starker Erntebelastungen, wobei SSR-Marker erheblichen Polymorphismus und geringe Inzucht aufzeigen. Die Art weist eine signifikante genetische Differenzierung zwischen nördlichen und südlichen Populationen auf, die durch Unterschiede im Lebensraum beeinflusst wird. Die Naturschutzmaßnahmen sollten sich auf die Erhaltung der genetischen Vielfalt durch ex-situ-Kultivierung, Wiederansiedlungsprogramme und die Bildung der lokalen Gemeinschaft konzentrieren, während auch grenzüberschreitende Naturschutzstrategien in Betracht gezogen werden sollten.
Referenz
- Senkoro AM, Talhinhas P, Simões F, Batista-Santos P, Shackleton CM, Voeks RA, Marques I, Ribeiro-Barros AI. Das genetische Erbe von Fragmentierung und Übernutzung im bedrohten medizinischen afrikanischen Pfefferbaum. Warburgia salutaris. Wissenschaftliche Berichte2020, 10(1):19725.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Pilze: Freunde oder Feinde – eine wissenschaftliche Outreach-Initiative zur Sammlung von luftgetragenen Pilzsporen durch Schüler der Oberstufe
Zeitschrift: Zeitschrift für Mikrobiologie und Biologiedidaktik
Jahr: 2024
Kleine, aber bedeutende genetische Differenzierung zwischen Populationen von Phyllachora maydis im Mittleren Westen der Vereinigten Staaten, aufgedeckt durch Mikrosatelliten (SSR)-Marker.
Journal: bioRxiv
Jahr: 2023
Das genetische Erbe von Fragmentierung und Übernutzung im bedrohten medizinischen afrikanischen Pfefferbaum, Warburgia salutaris
Journal: Wissenschaftliche Berichte
Jahr: 2020
Bewertung von Plasma-Biomarkern für die A/T/N-Klassifikation der Alzheimer-Krankheit bei Erwachsenen karibisch-hispanischer Ethnizität
Journal: JAMA Netzwerk Open
Jahr: 2023
Mehr ansehen Artikel, die von unseren Kunden veröffentlicht wurden.


