
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Variantenerkennung
Was ist Variant Calling?
Genetische Variation ist eine Art von Variation, die an Nachkommen vererbt werden kann und durch Veränderungen im genetischen Material eines Organismus verursacht wird. Diese Variation führt dazu, dass Organismen genetische Vielfalt auf unterschiedlichen Ebenen zeigen. Genetische Vielfalt ist die materielle Grundlage für das Überleben und die Entwicklung der menschlichen Gesellschaft und der Pflanzen. Es gibt viele Arten von genetischer Variation, von mikroskopisch sichtbaren Chromosomeninversionen bis hin zu einzelnen Nukleotidmutationen. Mit der Entwicklung der Genomik ist die Information über genetische Variation umfassender geworden und hat Folgendes eingeschlossen SNPInDel-, SV-, CNV- und Transposon-Mutationen, u. a.
Die Variantenbestimmung bezieht sich auf die Verwendung von Hochdurchsatzsequenzierung Technologie zur Sequenzierung und Analyse der Unterschiede im gesamten Genom eines Individuums oder einer Population einer Art, um eine große Menge an Informationen über genetische Variation zu erhalten, wie zum Beispiel Einzelnukleotid-Polymorphismus (SNP)Einfüge- und Löschstellen (InDel) sowie Stellen struktureller Variationen (SV), Kopienzahlvariationen (CNV) und weitere Informationen. Die Variantenbestimmung kann die grundlegendste und umfassendste Datenbasis für die anschließende funktionale Feinkartierung von Genen bereitstellen und schnell, genau und effizient die Unterschiede zwischen Genomen analysieren, jede Base des gesamten Genoms untersuchen und die umfangreichsten molekularen Marker erhalten.
Methoden zur Erkennung von strukturellen Varianten
Die Erkennung von strukturellen Varianten (SVs) ist ein wesentlicher Aspekt der genomischen Forschung. Verschiedene Methoden wurden entwickelt, um diese Aufgabe zu erfüllen, jede mit ihren spezifischen Stärken und Anwendungen:
- Read-Paar (RP) Methode
Die Read-Pair-Methode umfasst die Analyse von gepaarten Sequenzierungslesungen, um ihre räumlichen Beziehungen zu untersuchen. Strukturelle Varianten werden aus Anomalien in den erwarteten Einfügungsgrößen oder aus Inkonsistenzen in der Orientierung der gepaarten Lesungen abgeleitet. Abweichungen von den erwarteten Abständen oder Orientierungen deuten auf das Vorhandensein struktureller Varianten hin.
- Split-Read (SR) Methode
Die Split-Read-Methode konzentriert sich auf Fälle, in denen ein Segment eines Reads auf das Referenzgenom abgebildet wird, während sein Gegenstück dies nicht tut. Diese Technik ist besonders gut darin, Insertionen, Deletionen und andere komplexe strukturelle Veränderungen im Genom zu identifizieren. Die Methode zeichnet sich darin aus, präzise Bruchstellen von SVs zu bestimmen.
- Read-Depth (RD) Methode
Die Read-Depth-Methode bewertet die Tiefe der Sequenzierungsreads, die auf spezifische genomische Regionen abgebildet sind, um CNVs aufzudecken. Schwankungen in der Read-Tiefe können auf Erhöhungen oder Verringerungen der genomischen Kopienzahl hinweisen und somit CNVs offenbaren. Diese Methode ist vorteilhaft für die Erkennung von großflächigen Duplikationen oder Deletionen.
- Montagemethode (AS)
Die Versammlungsmethode nutzt Sequenzierungstechnologien der dritten Generation, wie PacBio SMRT oder Oxford Nanopore, in Verbindung mit de novo Assemblierungsansätzen, um umfangreiche und komplexe strukturelle Varianten zu identifizieren. Diese Methodik ermöglicht die Charakterisierung von langreichweitigen und komplexen genomischen Veränderungen direkt aus Rohsequenzierungsdaten und bietet einen umfassenden Überblick über SVs.
Vorteile und Merkmale der Variantenbestimmung
- Fülle: Detaillierte Analyse aller Aspekte genetischer Variation, einschließlich SNP, InDel, SV, SNV, neuartiges Gen usw.
- Flexibilität: mit oder ohne Bezug ist geeignet
- Genauigkeit: Verschiedene Sequenzierungsmethoden können je nach Material angewendet werden.
Anwendungen der Variantenaufrufung
- Krankheitsforschung: Die Entdeckung genetischer Varianten, die mit Krankheiten assoziiert sind, beleuchtet deren genetische Grundlagen und hilft, potenzielle Biomarker und Behandlungsziele zu identifizieren.
- Personalisierte Medizin: Genetische Erkenntnisse ermöglichen personalisierte Behandlungspläne und eine verbesserte Arzneimittelverwendung, indem sie Varianten identifizieren, die den Arzneimittelstoffwechsel beeinflussen.
- Agrarische Forschung: Die Identifizierung genetischer Varianten, die mit wünschenswerten Eigenschaften verbunden sind, hilft bei der Entwicklung neuer Sorten und der Steigerung der Produktivität in der Pflanzen- und Tierzucht.
Variant-Calling-Workflow

Dienstspezifikationen
Beispielanforderungen
|
|
 Klicken |
Sequenzierungsstrategie
|
 |
Bioinformatikanalyse Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an:
|
Sequenzierungstechnologie-Pipeline
- Basierend auf Whole-Genome-Assemblierung
- Basierend auf Whole-Genome-Resequenzierung
- Basierend auf der reduzierten Repräsentationsgenomsequenzierung
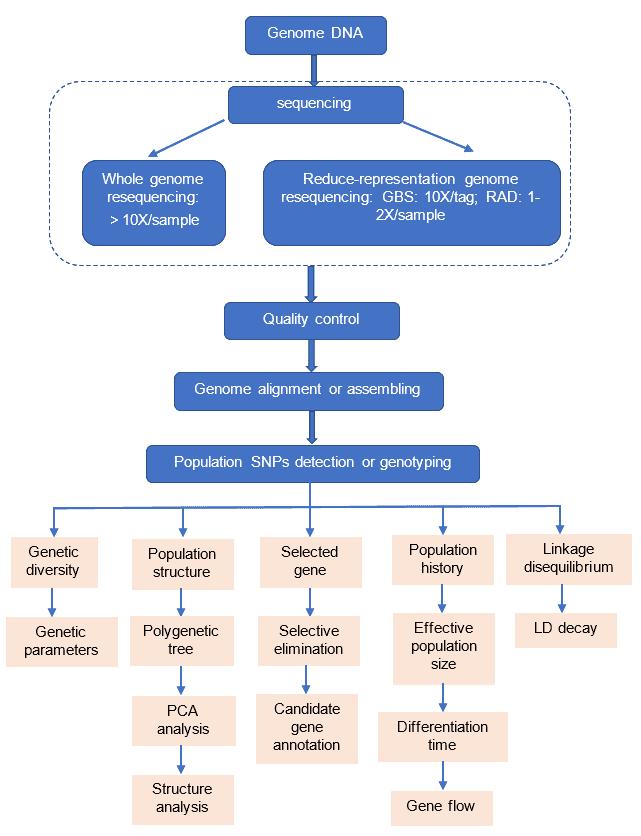
Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
Referenzen
- Jansen S, Aigner B, Pausch H, et al. Bewertung der genomischen Variation in einer Rinderpopulation durch Nachsequenzierung von Schlüssel-Tieren mit niedriger bis mittlerer Abdeckung. BMC Genomics, 2013, 14(1): 1.
- Zheng L Y, Guo X S, He B, et al. Genomweite Muster genetischer Variation in Zucker- und Körnersorghum (Sorghum bicolor). Genomik Biologie, 2011, 12(11):287-302.
- Alkan C, Coe BP, Eichler EE. Entdeckung und Genotypisierung von struktureller Variation im Genom. Naturwissenschaftliche Rezensionen GenetikMai 2011;12(5):363-76.
Demonstrationsergebnisse
Teilweise Ergebnisse sind unten aufgeführt:

Häufig gestellte Fragen zur Variantenbestimmung
1. Welche Arten von genetischen Varianten können nachgewiesen werden?
Genetische Varianten können grob in Sequenzvarianten, wie SNPs und kleine InDels, und strukturelle Varianten, einschließlich großer Deletionen, Duplikationen, Inversionen und Translokationen, unterteilt werden. Kopienzahlvariationen (CNVs) sind eine Unterart der SVs und können ebenfalls nachgewiesen werden.
2. Wie profitieren Long-Read-Sequenzierungstechnologien von der Variantenbestimmung?
Langzeit-Sequenzierung Technologien, wie PacBio SMRT-Sequenzierung und Oxford Nanoporen-Sequenzierung, bieten Vorteile wie die Erkennung von Varianten in komplexen genomischen Regionen, die für Methoden mit kurzen Reads herausfordernd sind. Sie bieten hohe Präzision, vermeiden PCR-Bias und ermöglichen die Auflösung der Variantenphasen.
3. Wie sieht der typische Arbeitsablauf für die Variantenbestimmung bei CD Genomics aus?
Der Arbeitsablauf umfasst die Datenverarbeitung und Qualitätskontrolle, das Zuordnen von Reads zum Referenzgenom, das Durchführen von Variantenaufrufen, das Annotieren von Varianten und das Visualisieren der Ergebnisse. Dies gewährleistet eine umfassende und genaue Erkennung genetischer Varianten.
Fallstudien zur Variantenbestimmung
Zusammenstellung des Genoms des nicht-kopfenden Pak Choi und Vergleich mit den Genomen des kopfenden Chinakohls und des Ölsaat-Raps.
Journal: Pflanzenbiotechnologie-Journal
Impactfaktor: 10,1
Veröffentlicht: 07. Dezember 2020
Hintergrund
Brassica-Kulturen umfassen diploide Arten und amphidiploide Arten. Das Genom von Pak Choi, das eng verwandt ist mit gelbem Sarson und Kopfkohl, wurde unter Verwendung von PacBio Einzelmolekül-Sequenzierung und Hi-C-Technologien. Diese Zusammenstellung identifizierte zahlreiche genetische Varianten, einschließlich SNPs, Indels und artspezifischer Gene, und vertiefte unser Verständnis der Vielfalt und der genetischen Merkmale von Brassica-Kulturen.
Materialien & Methoden
Probenvorbereitung
- Pflanzenmaterialien
- Blattproben
- DNA-Extraktion
Sequenzierung
- Genomsequenzierung
- PacBio SMRT-Sequenzierung
- RNA-Seq
- De novo Versammlung
- Chromosomenassemblierung und -bewertung
- Transkriptom-Analysen
- Wiederhole Annotation
- Genvorhersage
- funktionale Annotation
- Analyse struktureller Variationen
- Vergleichende Genomanalyse
Ergebnisse
In einer Online-Publikation aus dem Jahr 2020 über "Kohl"[1]Eine vergleichende Analyse der Variationen zwischen drei Kohlsorten, einschließlich der Variationen in der Anwesenheit/Abwesenheit von Genen (PAVs), strukturellen Varianten (SV) im Genom und Einzelne Nukleotid-Polymorphismen (SNPs), offenbarte bedeutende Erkenntnisse. Insbesondere wurden Unterschiede in den SV innerhalb der Gene für Blattform (z. B. KAN aus der GARP-Transkriptionsfaktor-Familie) und die hemmenden Rollen von blühbezogenen Genen (z. B. MAF4, SVP) unter den drei morphologischen Typen beobachtet (siehe Abbildung 1, links). Diese Erkenntnisse sind von entscheidender Bedeutung für das Verständnis der molekularen Mechanismen, die die Organmorphogenese und Blüte steuern, sowie für genetische Verbesserungen bei Kohl und verwandten Gemüsepflanzen.
 Abbildung 1: PAV und spezifische SVs im Kohlgenom[1]
Abbildung 1: PAV und spezifische SVs im Kohlgenom[1]
Andere Fälle:
Der Einfluss von strukturellen Varianten (SVs) auf die Genstruktur und Genexpression
Um die Anwendbarkeit der SV-Analyse zu validieren und ihren Nutzen in der evolutionären Populationsforschung zu erkunden, wurde 2020 eine Online-Publikation über "Tomaten" veröffentlicht.[2] wurde eine Untersuchung durchgeführt. Die Forscher erhielten einen systematischen evolutionären Baum basierend auf über 800 Materialien mit SNPs aus Daten der Kurzlesesequenzierung (Abbildung 2A). Anschließend wählten sie eine repräsentative Gruppe von 100 Materialien aus 7 Linien aus und sammelten Daten der Langlesesequenzierung, um ein Baumdiagramm basierend auf SV-Daten zu erstellen (Abbildung 2B).
Die Ergebnisse zeigten, dass die ausgewählten Materialien innerhalb ihrer bekannten taxonomischen Gruppen verteilt waren, was mit der SNP-basierten Klassifikation übereinstimmt. Dies zeigt die Eignung von SVs für die genetische Populationsanalyse.
 Abbildung 2: Systematischer evolutionärer Baum der Tomate[2]
Abbildung 2: Systematischer evolutionärer Baum der Tomate[2]
In einer Online-Publikation aus dem Jahr 2020 über "Reis"[3]Forscher stellten Diagramme zur Populationsstruktur basierend auf sowohl SNP-Daten (Abbildung 3A) als auch SV-Daten (Abbildung 3B) zusammen. Die SNP-Analyse zeigte eine grobe Unterteilung der Population in etwa sechs Gruppen, mit einer klaren Unterscheidung zwischen japonica- und indica-Reissorten. Interessanterweise lieferte die SV-Analyse hochgradig konsistente Ergebnisse, die die in der SNP-Analyse beobachtete Differenzierung weiter unterstützten.
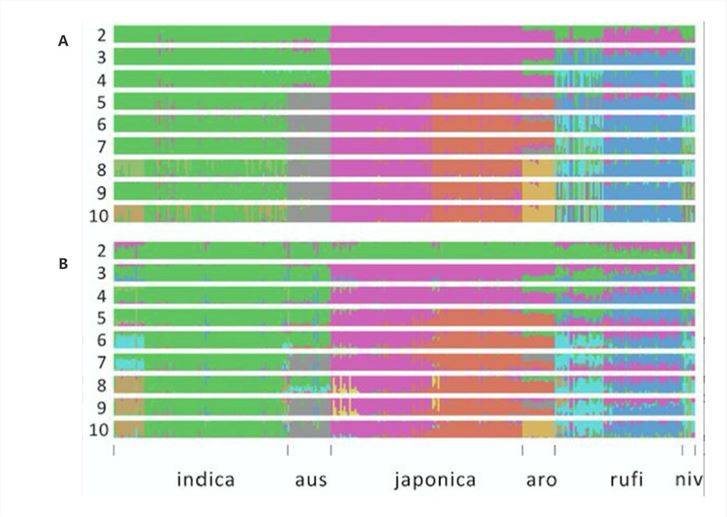 Abbildung 3: Genetische Struktur von Reis[3]
Abbildung 3: Genetische Struktur von Reis[3]
Bevölkerung SV-Mutationsfrequenzspektrum
Um die erheblichen Auswirkungen schädlicher Varianten zu untersuchen, die Veröffentlichung von 2020 über Reis[3] berechnete Frequenzspektren für nicht-kodierende Stellen in verschiedenen Gruppen (Abbildung 4-A, B, C). Jede SFS umfasst fünf SV-Typen (DUP, DEL, TRA, MEI und INV) sowie zwei SNP-Typen (Syn, Nsyn) und zeigt drei bemerkenswerte Merkmale:
Es bestehen signifikante Unterschiede zwischen verschiedenen Populationen, die mit einem verstärkten genetischen Drift während der Flaschenhals-Effekte der Domestikation und Veränderungen in den Fortpflanzungssystemen übereinstimmen.
Der Anteil fester SVs ist geringer als der fester synonymer SNPs und nicht-synonymer SNPs. SVs haben eine niedrigere Häufigkeit im Genom und werden nach Mutationen eher beseitigt, was auf eine höhere Wahrscheinlichkeit von Schäden im Zusammenhang mit SVs hinweist.
INV-Ereignisse zeigen die extremsten SFS, wobei über 90 % der INV-Ereignisse in drei oder weniger Individuen in jeder Gruppe identifiziert werden, was darauf hindeutet, dass INV-Ereignisse während des evolutionären Prozesses möglicherweise einer starken Selektion unterliegen.
Forscher führten eine statistische Analyse der Standorte von SVs und SNPs auf den Chromosomen durch, und die Testergebnisse zeigen eine signifikante Korrelation zwischen der Vielfalt der SVs und SNPs in chromosomalen Fenstern (Abbildung 4-D). Dies deutet darauf hin, dass SVs genetische Informationen über Populationen liefern, die grundsätzlich mit SNPs übereinstimmen.
 Abbildung 4: SV-Frequenzspektren[3]
Abbildung 4: SV-Frequenzspektren[3]
Linkage-Disequilibrium-Analyse mit SVs
In der [Reis] Studie[3]LD (Linkage Disequilibrium) wurde für drei verschiedene Populationen unter Verwendung von SNP-, SV- und SNP+SV-Daten berechnet. Aufgrund ihrer potenziell schädlichen Auswirkungen weisen SVs oft niedrigere Populationsfrequenzen im Vergleich zu SNPs auf und können eine schnellere LD-Zerfall über physische Distanzen erfahren (Abbildung 5). Die SNP-Daten zeigten, dass innerhalb von etwa 100 kb der r2 für japonica SNPs bei etwa 0,2 blieb, während er für indica ungefähr 0,1 betrug. In derselben physischen Distanz lag der r2 für rufipogon unter 0,05. Die r2-Werte für SVs waren jedoch in allen Populationen niedriger als die für SNPs, wobei Werte von über 0,1 nur innerhalb sehr kurzer Distanzen (<15 kb) erreicht wurden.
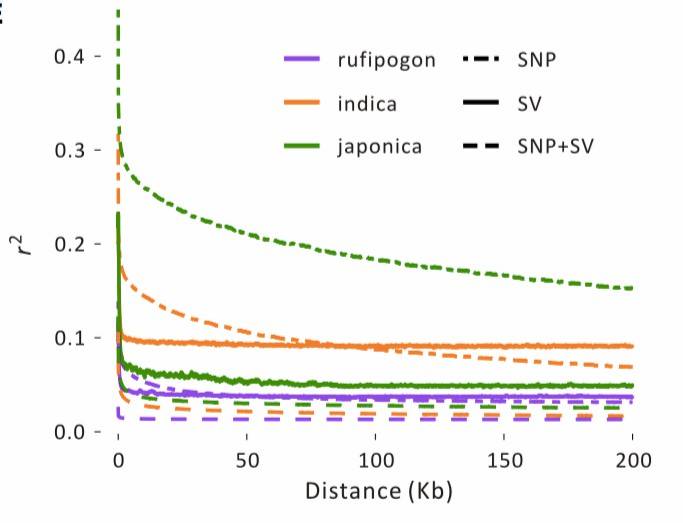 Abbildung 5: Analyse des Linkage-Disequilibriums[3]
Abbildung 5: Analyse des Linkage-Disequilibriums[3]
Studie zur Domestikation von Populationen
In der durchgeführten Forschung zu Reis[3]Eine Bewertung der genomischen Unterschiede zwischen Einzel-Nukleotid-Polymorphismen (SNPs) und strukturellen Varianten (SVs) zeigte eine bemerkenswerte Unterscheidung. Die Studie stellte fest, dass die durchschnittliche FST-Schätzung für SNPs im Vergleich zu SVs deutlich höher war. Dieses Ergebnis deutet darauf hin, dass SVs typischerweise niedrigere Populationsfrequenzen im Vergleich zu SNPs aufweisen.
Durch die Integration dieser Daten mit gut etablierten Domestikations- und Verbesserungsgenen bestätigte die Untersuchung die erhebliche Anreicherung dieser Gene innerhalb der obersten 1% und 10% FST-Intervalle. Dies lieferte wertvolle Einblicke in funktionale Gene, die mit physiologischen Prozessen, morphologischen Merkmalen und Lebensmittelqualität assoziiert sind (Abbildung 3).
 Abbildung 6: SV-Eigenschaften im Zusammenhang mit der Domestikation[3]
Abbildung 6: SV-Eigenschaften im Zusammenhang mit der Domestikation[3]
Whole-Genome-Assoziationsanalyse
In der Veröffentlichung von 2020 über [Raps][4]Eine GWAS (Genome-Wide Association Study) wurde unter Verwendung der identifizierten PAVs aus acht Rapsvarianten durchgeführt. Diese Analyse zeigte ursächliche Zusammenhänge zwischen PAVs und Merkmalen wie Schotenlänge, Samengewicht und Blütezeit. Interessanterweise wurden diese signifikanten Entdeckungen in den SNP-GWAS-Ergebnissen übersehen (Abbildung 7).
Ähnlich in der Veröffentlichung von 2021 über [Pfirsich][5]Es wurden Kandidaten-SVs beobachtet, die mit Eigenschaften wie frühem Fruchtreifen, Perikarpfarbe um den Stein, Fruchtform und flacher Formbildung assoziiert sind (Abbildung 8).
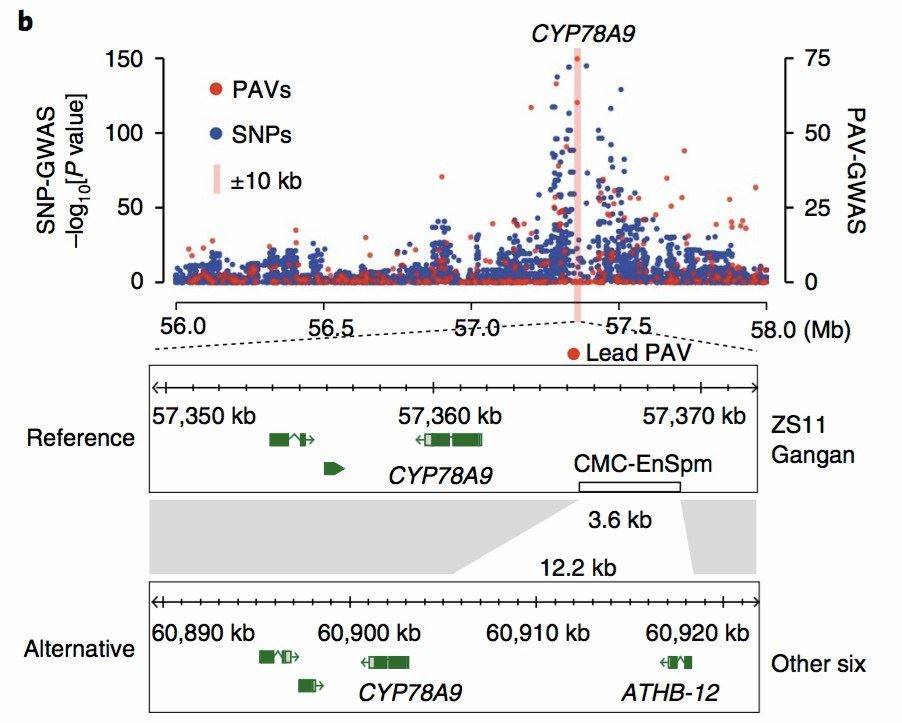 Abbildung 7: Ergebnisse der PAV_GWAS-Analyse bei Raps[4]
Abbildung 7: Ergebnisse der PAV_GWAS-Analyse bei Raps[4]
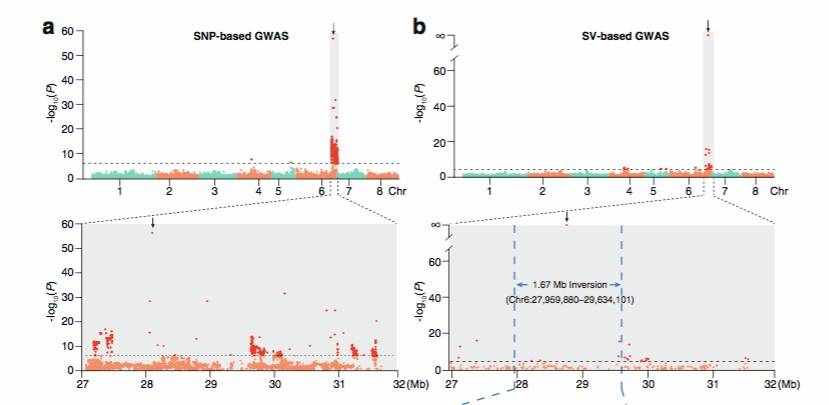 Abbildung 8: Ergebnisse der SV_GWAS-Analyse bei Pfirsichen[5]
Abbildung 8: Ergebnisse der SV_GWAS-Analyse bei Pfirsichen[5]
Referenzen
- Li P, Su T, Zhao X, et al. Zusammenstellung des Genoms von nicht-kopfendem Pak Choi und Vergleich mit den Genomen von kopfendem Chinakohl und dem Ölsaat-Raps. Pflanzenbiotechnologie Journal, 2020.
- Alonge M, Wang X, Benoit M, et al. Wesentliche Auswirkungen weit verbreiteter struktureller Variationen auf die Genexpression und die Verbesserung von Nutzpflanzen bei Tomaten. Zelle, 2020.
- Yixuan K, Yi L, Tuomas T, et al. Evolutionäre Genomik struktureller Variation bei der Domestikation von asiatischem Reis (Oryza sativa). Molekulare Biologie und Evolution, 2020.
- Song J, Guan Z, Hu J, et al. Acht hochqualitative Genome enthüllen die Pan-Genom-Architektur und die Ökotyp-Differenzierung von Brassica napus. Natur Pflanzen, 2020.
- Jiantao G, Yaoguang X, Yang Y, et al. Analysen der Genomstrukturvariationen von Pfirsichen zeigen die Populationsdynamik und eine 1,67 Mb große kausale Inversion für die Fruchtform. Genom Biologie, 2021.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Sammlung genetischer Daten in ethnisch basierten Studien bei Aymaras, Quechuas und Mestizen: die Herausforderungen der Genetik von Alzheimer in der peruanischen Bevölkerung (GAPP) Studie
Zeitschrift: Alzheimer & Demenz
Jahr: 2022
Bewertung von Plasma-Biomarkern für die A/T/N-Klassifikation der Alzheimer-Krankheit bei Erwachsenen karibischer hispanischer Ethnie
Zeitschrift: JAMA Network Open
Jahr: 2023
Erhöhte Produktion von pathogenen, luftgetragenen Pilzsporen bei der Exposition einer Bodenmykobiota gegenüber chlorierten aromatischen Kohlenwasserstoffschadstoffen
Journal: Mikrobiologie Spektrum
Jahr: 2023
Eine Splice-Variante im SLC16A8-Gen führt zu einem Defizit beim Laktattransport in aus menschlichen iPS-Zellen abgeleiteten retinalen Pigmentepithelzellen.
Journal: Zellen
Jahr: 2021
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


