
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Kleine RNA-Sequenzierung
Um das wachsende Forschungsinteresse an kleinen RNAs zu unterstützen, bietet CD Genomics einen qualifizierten Service für die Sequenzierung kleiner RNAs an, der die Entdeckung neuer kleiner RNAs, die Charakterisierung von Mutationen und die Expressionsprofilierung kleiner RNAs durch den Einsatz fortschrittlicher Technologien abdeckt. NGS-Technologien und Datenanalyse-Pipeline.
Die Einführung der kleinen RNA-Sequenzierung
Kleine RNA-Spezies umfassen in der Regel die häufigsten und am besten untersuchten. MikroRNA (miRNA)kleine interferierende RNA (siRNA) und piwi-interagierende RNA (piRNA) sowie andere Arten von kleinen RNAs, wie kleine nukleolare RNA (snoRNA) und kleine nukleare RNA (snRNA). Kleine RNA ist eine Art von geringfügig vorkommenden, kurzen RNA (<200 nt), die nicht für Proteine kodieren und keine Polyadenylierung aufweisen. Die Populationen kleiner RNAs können zwischen verschiedenen Gewebetypen und Arten erheblich variieren. Im Allgemeinen entstehen kleine RNAs durch Fragmentierung längerer RNA-Sequenzen mit Hilfe spezieller Enzymgruppen und anderer Proteine.
Kleine RNAs wirken bei der Genstilllegung und der posttranskriptionalen Regulation der Genexpression. Allerdings ist kleine RNA nicht ausreichend für die Induktion der RNA-Interferenz. Sie muss in der Regel den Kern des RNA-Protein-Komplexes bilden, der als RNA-induziertes Silencing-Komplex (RISC) bekannt ist. siRNAs können die mRNA in der Mitte des mRNA-siRNA-Duplexes spalten, und die resultierenden mRNA-Hälften werden von anderen zellulären Enzymen abgebaut. Im Gegensatz zum siRNA-Weg wird der Abbau von miRNA-vermittelter mRNA durch die enzymatische Entfernung des polyA-Schwanzes der mRNA eingeleitet. piRNAs sind entscheidend für die Entwicklung von Keimzellen. Es wurde nachgewiesen, dass kleine RNAs an einer Reihe biologischer Prozesse beteiligt sind, einschließlich Entwicklung, Zellproliferation und -differenzierung sowie Apoptose.
Durch die Nutzung einer enormen Leistung mit beispielloser Empfindlichkeit und Dynamikbereich, NGS kann schwach exprimierte kleine RNAs identifizieren und gleichzeitig die Heterogenität in Länge und Sequenz quantitativ aufzeigen. NGS ist ein leistungsstarkes Werkzeug zur Untersuchung der Funktion von kleinen RNAs und zur Vorhersage potenzieller mRNA-Zielmoleküle, ohne dass verfügbare Referenzgenome erforderlich sind. Der Erwerb einer hochwertigen kleinen RNA-Sequenzierungsbibliothek beginnt mit der Isolation kleiner RNAs durch Größenfraktionierung mittels Gel-Elektrophorese oder Silica-Spin-Säulen aus totaler RNA. Nach der RNA-Adapter-Ligation unter Verwendung eines 5'-adenylierten DNA-Adapters mit einem blockierten 3'-Ende werden kleine RNAs revers transkribiert, durch PCR amplifiziert und sequenziert. Um bekannte miRNAs zu identifizieren und zu annotieren, können die Sequenzierungsreads einer artspezifischen Datenbank, wie mirWalk und miRBase, zugeordnet werden.
Vorteile unseres Small RNA-Sequenzierungsdienstes
- Kleine RNA und miRNA-Profiling
- Verstehen, wie die posttranskriptionale Regulation zum Phänotyp beiträgt
- Identifizierung weiterer nicht kartierter kleiner RNAs und Isoformen sowie neuer Biomarker
- Hohe Auflösung, hohe Genauigkeit: Erkennung von Einzel-Nukleotid-Unterschieden, präzises Zählen von wenigen bis zu zehntausenden Kopien
- Hoher Durchsatz, hohe Qualität: Über 8 Millionen Sequenzen in einem Sequenzierungslauf erhalten, kurze Versuchszeit, hohe Effizienz, niedrige Kosten und zuverlässige Qualität.
- Standardisierter analytischer Workflow: Professionelle Bioinformatik-Teams und Analyse-Dienste zur Bereitstellung standardisierter, fortschrittlicher und maßgeschneiderter Analysen.
- Umfangreiche Erfahrung in der kleinen RNA-Sequenzierung und -Analyse: Professionelle Sequenzierungstechniker unterstützen bei der experimentellen Planung, Problemlösung und Ergebnisanalyse.
- Angepasste kleine RNA-Sequenzierungsprotokolle: Personalisierte kleine RNA-Sequenzierungspläne entsprechend spezifischer Bedürfnisse anpassen.
Kleines RNA-Sequenzierungs-Workflow
CD Genomics nutzt die Illumina HiSeq-Plattformen zur Sequenzierung von kleinen RNAs. Wir haben flexible Strategien zur Entdeckung und Profilierung von miRNA (15-30 nt) und/oder kleinen RNAs (30-200 nt). Unser hochqualifiziertes Expertenteam führt ein Qualitätsmanagement durch, das jede Prozedur überwacht, um zuverlässige und unvoreingenommene Ergebnisse zu gewährleisten. Der allgemeine Arbeitsablauf für die Sequenzierung von kleinen RNAs ist unten skizziert.

Dienstspezifikation
Musteranforderungen
|
|
 Klicken |
Sequenzierungsstrategien
|
 |
Datenanalyse Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an:
|
Analyse-Pipeline
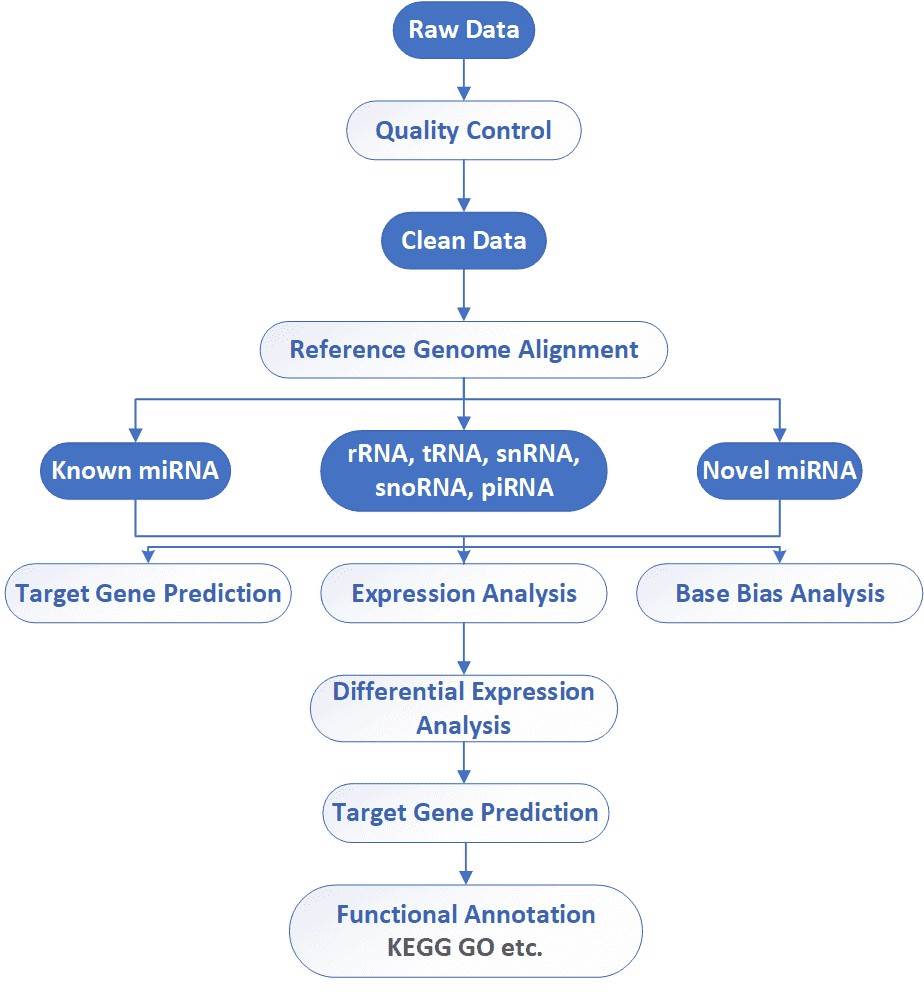
Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details zur kleinen RNA-Sequenzierung für Ihre Schreibanpassung.
CD Genomics bietet umfassende Dienstleistungspakete für die kleine RNA-Sequenzierung an, die die Standardisierung von Proben, die Bibliotheksvorbereitung, die Tiefensequenzierung, die Qualitätskontrolle der Rohdaten, die Genomassemblierung und maßgeschneiderte bioinformatische Analysen umfassen. Wir können diese Pipeline an Ihr Forschungsinteresse anpassen. Wenn Sie zusätzliche Anforderungen oder Fragen haben, zögern Sie bitte nicht, sich zu melden. Kontaktieren Sie uns.
Demo-Ergebnisse
Teilweise Ergebnisse sind unten aufgeführt:

Verteilung der Sequenzierungsqualität
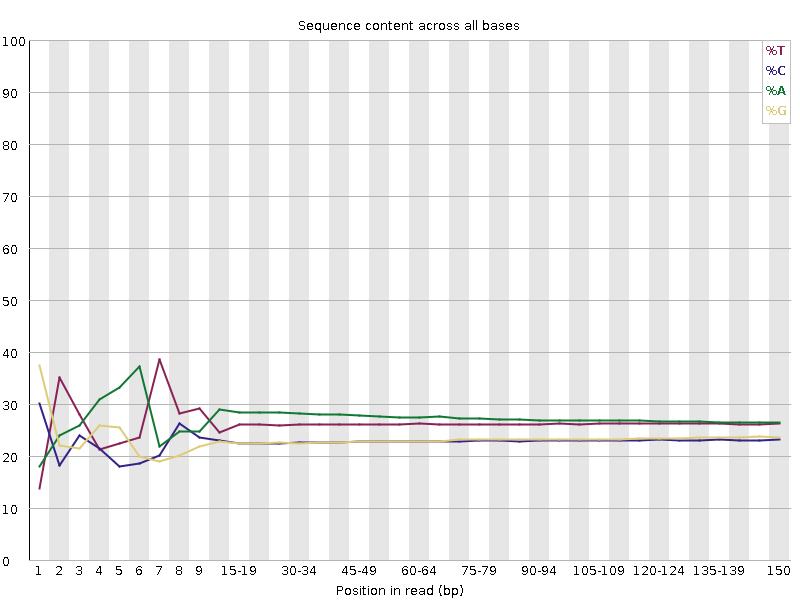
A/T/G/C-Verteilung
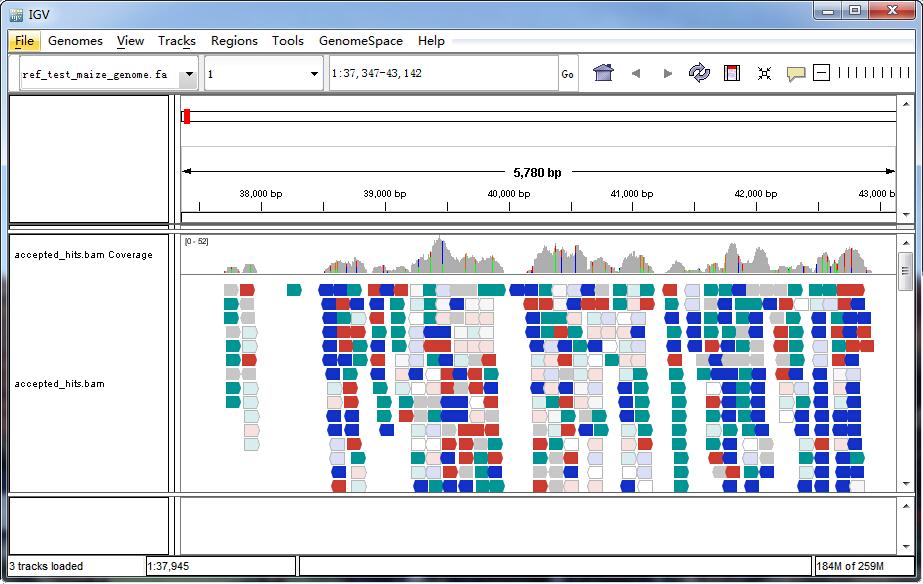
IGV-Browser-Oberfläche
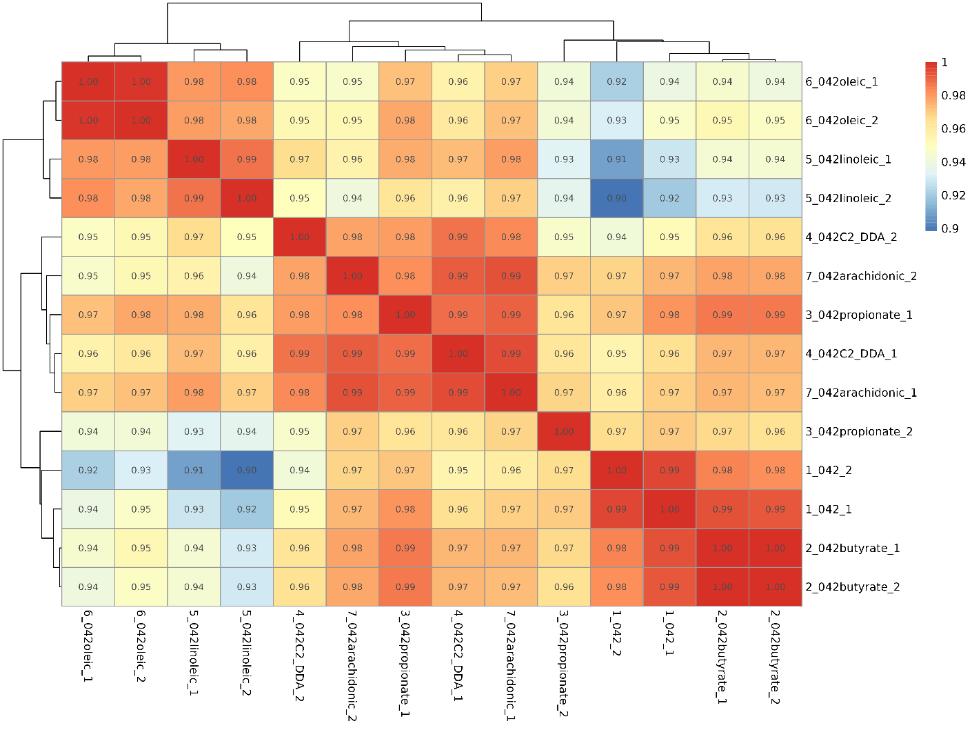
Korrelationsanalyse zwischen Proben
PCA-Score-Plot

Venn-Diagramm
Vulkan-Plot
Statistik Ergebnisse der GO-Annotation
KEGG-Klassifikation
Kleine RNA-Seq FAQs
1. Wie ist der Workflow für die Datenvorverarbeitung?
Die ersten Schritte sind das Trimmen der Adapter mit FASTX und eine optionale k-mer-Korrektur mit ECHO. Anschließend wird das Expressionsprofil durch Genom-Mapping und Annotation erstellt. Wenn die Anzahl der Reads pro Probe variiert, wird eine Normalisierung angewendet, um die Effekte der Unterstichprobenahme auszugleichen.
2. Wie viele Reads sind für die kleine RNA-Sequenzierung erforderlich?
Es hängt von Ihrer Anwendung ab. Für die Expressionsprofilierung liegt ein akzeptierter Bereich für gemappte Reads pro Probe bei 100K-2M. Für Entdeckungsanwendungen sollten mindestens 5-10M in Betracht gezogen werden.
3. Wie viele biologische Replikate benötige ich für jede Bedingung?
Biologische Replikate können das Vertrauen erhöhen und experimentelle Fehler reduzieren. Wir empfehlen, mindestens drei Replikate pro Probe einzureichen. Beachten Sie, dass die endgültige Anzahl der Replikate durch Ihre endgültigen experimentellen Bedingungen bestimmt wird.
4. Wie sollte ich Proben versenden?
Sie können mit Trockeneis oder RNAstable bei Raumtemperatur versenden. Empirisch ergibt der Versand von RNA-Proben in RNAstable eine gute RNA-Qualität nach der Rehydrierung.
5. Kann ich einreichen E. coli Zellen oder Gewebeproben?
Ja, neben RNAs können Sie auch gefrorene Proben einreichen. E. coli Zellpellets oder Gewebe mit Trockeneis.
6. Ist Poly-A-Anreicherung oder RNA-Depletion für die Mikrorna-Sequenzierung erforderlich?
Keine Anreicherung ist erforderlich für MikroRNA-Sequenzierung weil Adapter selektiv nur an kleine RNAs ligiert werden.
7. Ich stellte fest, dass ich die heruntergeladene Fastq-Datei nicht öffnen konnte.
Bitte stellen Sie sicher, dass Ihr Computer über genügend RAM verfügt, um die großen Dateien zu öffnen. Sie können sie mit einem Texteditor auf Windows-Computern mit ausreichend RAM oder auf Linux-Computern, was die bevorzugte Option ist, öffnen. Alternativ können Sie auch andere spezifische Fastq-Reader in Betracht ziehen.
Kleine RNA-Seq Fallstudien
Neuartige Entdeckung von Mikro-RNA durch kleine RNA-Sequenzierung im postmortalen menschlichen Gehirn
Journal: BMC Genomics
Impactfaktor: 3,729
Veröffentlicht: 4. Oktober 2016
Hintergrund
Mikro-RNAs (miRNAs) regulieren die Genexpression hauptsächlich durch die translationalen Repression von Ziel-mRNA-Molekülen. Über 2700 menschliche miRNAs wurden identifiziert, und einige wurden mit Krankheitsphänotypen in Verbindung gebracht und zeigten gewebespezifische Expressionsmuster. Die Autoren führten Studien zur Sequenzierung kleiner RNAs an 93 postmortalen Proben des präfrontalen Kortex von Patienten mit Huntington-Krankheit oder Parkinson-Krankheit durch. Dabei entdeckten sie erfolgreich 99 putative neuartige miRNAs.
Materialien & Methoden
- Gefrorenes Gehirngewebe
- Isolation und Reinigung von totaler RNA
- Bibliothekskonstruktion
- Einzelend-Sequenzierung
- Illumina HiSeq 2000 System
- miRNA-Sequenzierung
- Kleine RNA-Sequenzierung
- Identifizierung neuartiger miRNAs
- Sequenzähnlichkeit mit bekannten miRNAs
- Differenzielle Expressionsanalyse
Ergebnisse
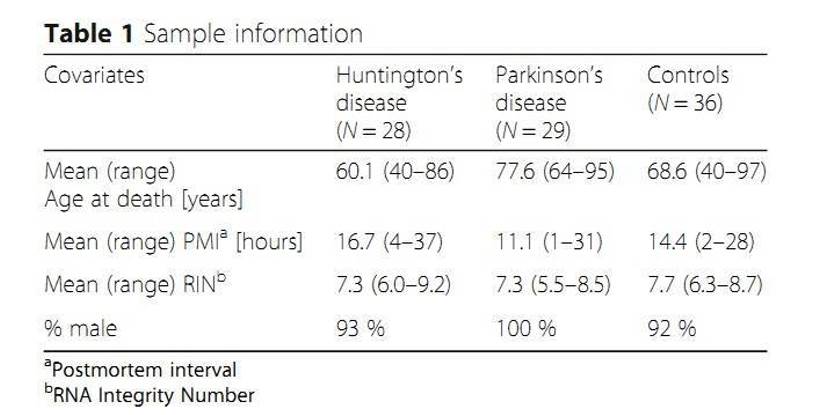
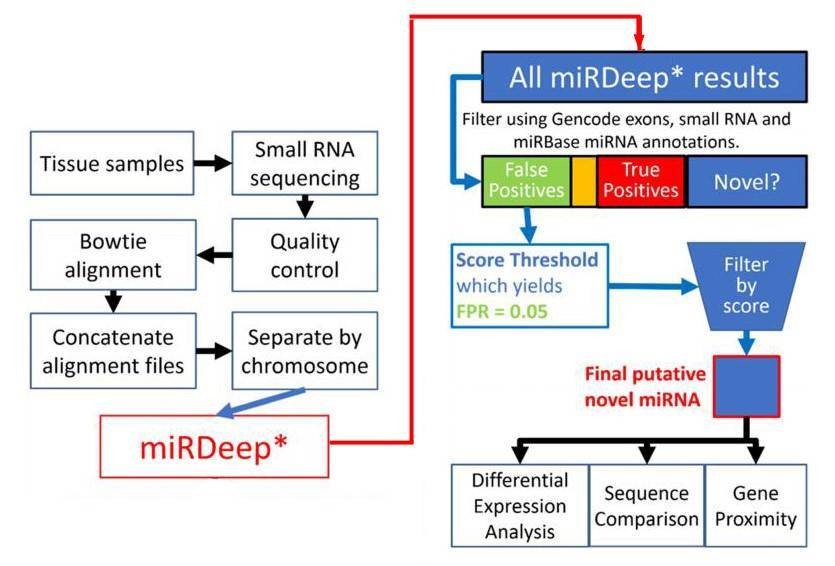 Abbildung 1. Flussdiagramm-Darstellung des neuartigen miRNA-Entdeckungsprozesses.
Abbildung 1. Flussdiagramm-Darstellung des neuartigen miRNA-Entdeckungsprozesses.
Insgesamt wurden 8891 miRNAs identifiziert.
Die miRDeep*-Analyse von Sequenzdaten aus 93 Proben des menschlichen präfrontalen Kortex identifizierte 8891 miRNAs. Nach der Filterung bekannter miRNAs aus miRBase v20, anderen kleinen RNAs und Exons blieben 3641 miRNAs übrig. Der Rest wurde anhand des miRDeep*-Scores gefiltert. Höhere Scores deuten auf eine höhere Zuverlässigkeit des miRNA-Ergebnisses hin. Insgesamt wurden schließlich 99 potenzielle neuartige miRNAs identifiziert.
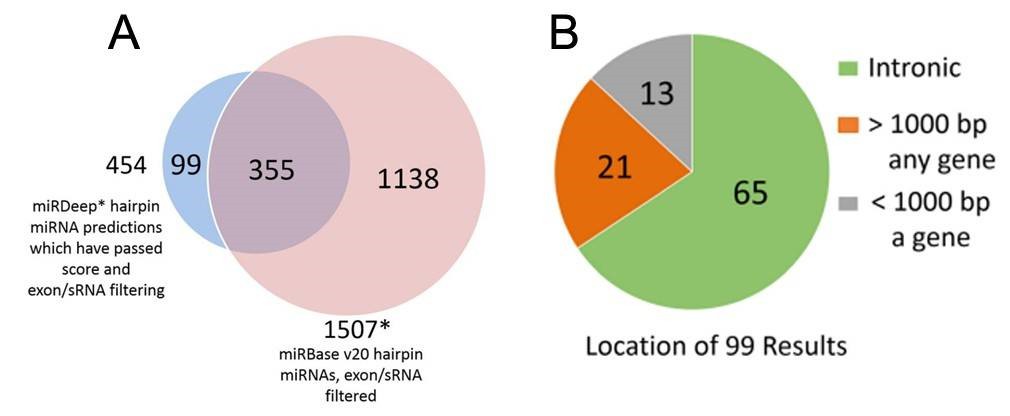 Abbildung 2. miRDeep* potenzielle miRNAs und miRBase miRNAs (A) sowie genomische Standorte der potenziellen neuartigen miRNAs (B).
Abbildung 2. miRDeep* potenzielle miRNAs und miRBase miRNAs (A) sowie genomische Standorte der potenziellen neuartigen miRNAs (B).
Insgesamt 99 vermeintlich neue miRNAs
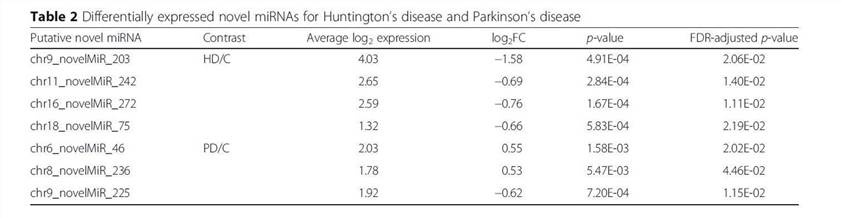
Mit dem SSEARCH-Ausrichter haben 34 der 99 vermeintlichen neuartigen reifen miRNAs gut mit mindestens einer reifen miRNA aus der miRBase v20 übereingestimmt. Die Analyse der differentiellen Expression unter Verwendung linearer Modelle, die das Alter zum Zeitpunkt des Todes berücksichtigen, zeigte, dass 4 der 99 vermeintlichen miRNAs differentiell zwischen Kontroll- und Huntington-Krankheitsproben exprimiert sind und dass 3 differentiell zwischen Kontroll- und Parkinson-Krankheitsproben exprimiert sind (Tabelle 2). Die detaillierten Daten sind in Tabelle 2 dargestellt, die von LIMMA v. 3.33.7 generiert wurden.
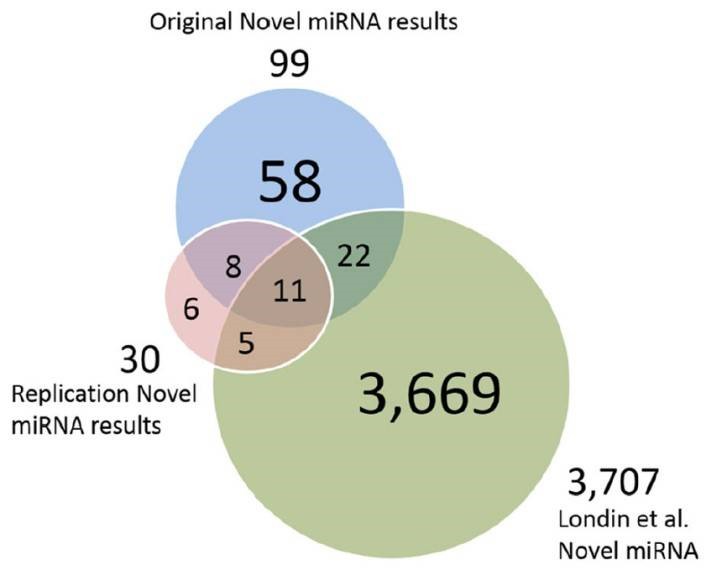 Abbildung 3. Vermutete neuartige miRNAs, Replikationsdaten miRNAs und Londin u. a.miRNAs.
Abbildung 3. Vermutete neuartige miRNAs, Replikationsdaten miRNAs und Londin u. a.miRNAs.
Fazit
Die Autoren entwickelten eine Pipeline unter Verwendung von miRDeep*, gepooltem RNA-Sequencing und Score-Filterung, um 99 vermeintlich neuartige miRNAs aus 93 postmortalen menschlichen Proben des präfrontalen Kortex zu entdecken. Sieben dieser miRNAs wurden experimentell validiert, und 19 wurden in einem unabhängigen Datensatz repliziert. Einige miRNAs zeigten eine unterschiedliche Expression zwischen HD- und Kontrollproben oder PD- und Kontrollproben, was auf potenzielle Rollen in neurodegenerativen Erkrankungen hindeutet. Diese Ergebnisse unterstreichen die Vielfalt der menschlichen miRNAs, insbesondere in gewebespezifischen Kontexten, und deren potenzielle Implikationen für das Verständnis von Erkrankungen wie HD und PD.
Referenz:
- Wake C, Labadorf A, Dumitriu A, u. a.Neuartige microRNA-Entdeckung mittels kleiner RNA-Sequenzierung im postmortalen menschlichen Gehirn. BMC Genomics, 2016, 17(1): 776.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Simultane Repression des Kohlenstoffkatabolismus steuert die gleichzeitige Nutzung von Zucker und aromatischen Verbindungen in Pseudomonas putida M2.
Zeitschrift: Angewandte und Umweltmikrobiologie
Jahr: 2023
IL-4 fördert die Erschöpfung von CD8.+ CART-Zellen
Zeitschrift: Nature Communications
Jahr: 2024
Fettreiche Diäten während der Schwangerschaft führen zu Veränderungen der DNA-Methylierung und Proteinexpression im Pankreasgewebe des Nachwuchses: Ein Multi-Omics-Ansatz
Internationale Zeitschrift für Molekulare Wissenschaften
Jahr: 2024
KMT2A assoziiert mit dem PHF5A-PHF14-HMG20A-RAI1 Subkomplex in Stammzellen des Pankreaskarzinoms und reguliert epigenetisch deren Eigenschaften.
Zeitschrift: Nature Communications
Jahr: 2023
Krebsassoziierte DNA-Hypermethylierung von Polycomb-Zielen erfordert die doppelte Erkennung von Histon H2AK119-Ubiquitinierung und der sauren Tasche des Nukleosoms durch DNMT3A.
Journal: Wissenschaftliche Fortschritte
Jahr: 2024
Genomisches Imprinting-ähnliches monoalleles väterliches Ausdrucksmuster bestimmt das Geschlecht von Kanalkatfischen.
Journal: Wissenschaftliche Fortschritte
Jahr: 2022
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


