
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben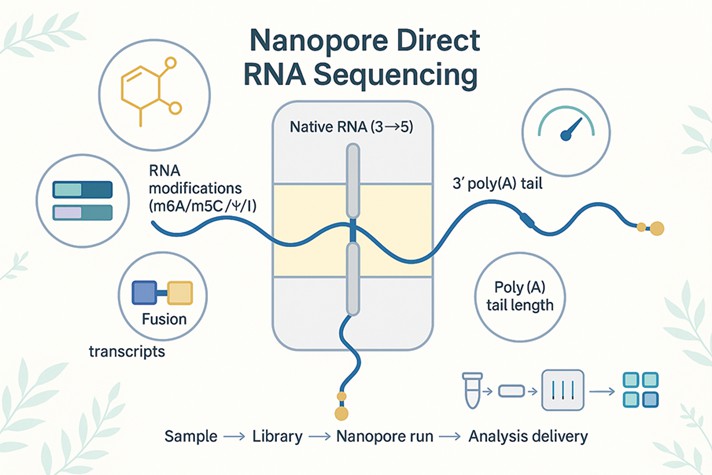
Überblick: Was ist die Nanopore-Direkt-RNA-Sequenzierung?
Nanopore-Direkt-RNA-Sequenzierung ist der einzige Ansatz, der native RNA-Moleküle direkt liest – ohne Reverse Transkription oder PCR – sodass Ihre Daten reale Basismodifikationen und molekularspezifische Merkmale bewahren. Die Sequenzierung erfolgt 3′→5′ von einem poly(T)-Adapter und erzeugt Einzelmolekül-Lesungen, die vollständige 5′-UTR–CDS–3′-UTR-Strukturen abdecken.
Wie es funktioniert
- Anreichern und anpassen: Poly(A)+ RNA (oder eine benutzerdefinierte Erfassung) wird vorbereitet und mit einem Motorprotein an einen Poly(T)-Adapter ligiert.
- Translokation durch die Pore: Jeder RNA-Strang wird durch eine Nanopore gezogen, wo Änderungen des ionischen Stroms die Nukleotididentitäten kodieren.
- Dekodierung und Analyse: Die Basiserkennung rekonstruiert die Sequenz, während signalbewusste Algorithmen RNA-Modifikationssignaturen (m6A/m5C/Ψ/I) und die Poly(A)-Schwanzlänge pro Lesevorgang ausgeben.
- Die Orientierung und Chemie sind in unserem validierten Nanopore-Protokoll für die direkte RNA-Sequenzierung dokumentiert, das Teil des Arbeitsablaufs für die direkte RNA-Sequenzierung mit Nanopore ist.
Warum es wichtig ist
- Vollständige Isoformen, keine Assemblierung: Lange Reads lösen direkt alternatives Splicing und komplexe Transkriptstrukturen auf, wodurch Inferenzfehler, die bei Kurz-Reads häufig auftreten, reduziert werden. RNA-Seq.
- Native Epitranskriptom: Da es keine RT/PCR gibt, bleiben echte chemische RNA-Modifikationen erhalten und sind mit nahezu basenaufer Auflösung nachweisbar.
- Poly(A)-Biologie: Die Poly(A)-Schwanzlänge pro Lesevorgang ermöglicht Studien zur Stabilität, Translation und isoformspezifischen Regulation.
- Geringere Amplifikation/GC-Bias: PCR-freie Bibliotheken verbessern die Quantifizierung von GC-extremen und strukturierten RNAs.
Nanopore Direkte RNA-Sequenzierung Arbeitsablauf
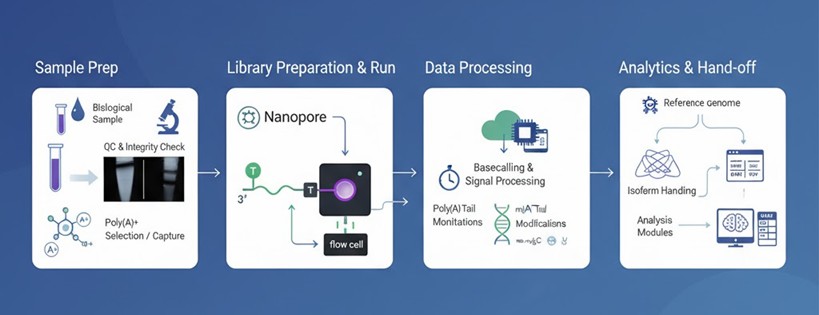
1. Musterbeleg & Qualitätskontrolle
- Akzeptierte Eingabe: Gesamt-RNA (empfohlen ≥50 µg bei ≥180 ng/µL; brauchbar 20–80 µg, studienabhängig).
- Integritätsprüfung (Bioanalyzer/TapeStation), Kontaminanten-Screen, rRNA-Gehalt; RNase-freie Handhabung.
- Optionale Kontrollen: ERCC-Spike-ins; Methylierungsstandards zur Kalibrierung des Anrufers.
- Empfehlung: ≥2 biologische Replikate pro Bedingung (≥3 verbessern die Aussagekraft).
2. Poly(A)+-Selektion / benutzerdefinierte Erfassung
- Standard: Oligo-dT-Anreicherung für mRNA.
- Optionen: rRNA-Depletion oder gezielte Erfassung (z. B. lncRNA/circRNA-Panels oder organismspezifische Sonden).
3. Bibliotheksvorbereitung — Direkte RNA 1D (PCR-frei)
- Ligation des Poly(T)-Adapters und Motorproteins; kein RT / keine PCR.
- Die Orientierung ist 3′→5′ nach Vorgabe; Chemie und Barcode-Registrierung sind in unserem validierten Direct RNA Sequencing Nanopore-Protokoll dokumentiert.
4. Nanopore-Lauf und Echtzeitüberwachung
- Flow-Cell-QC, Porenbelegung, Live-Ertragsverfolgung.
- Die Ausführungskonfiguration entspricht den Tiefenzielen (Organismengröße, Komplexität, Vergleiche).
5. Basisabgleich und Signalverarbeitung
- Rohes FAST5 → FASTQ mit Qualitätsmetriken.
- Signalbewusste Extraktion der Poly(A)-Schwanzlängen und Modifikationsmerkmale (m6A/m5C/Ψ/I-Kandidaten) pro Lesevorgang.
6. Ausrichtung und Isoformverarbeitung
- Referenzgeführte gespleißte Ausrichtung.
- Isoform-Clustering und De-Duplizierung zur Minderung von 5'-Trunkierung und Molekülvielfalt; Nutzung von Poly(A)-Stellen erfasst.
7. Primäre Analyseübergabe
- Strukturierte Ausgaben (BAM/CRAM, GTF/GFF, pro-Lese Poly(A)-Tabellen, Modifikationsmerkmals-Tabellen) wurden in die Nanopore-Analysemodule für die direkte RNA-Sequenzierung übergeben.
Hinweise & bewährte Praktiken
- PCR-freie Bibliotheken reduzieren GC-/Amp-Bias und bewahren native RNA-Modifikationen.
- Eine gewisse 5′-Trunkierung wird erwartet; unsere Cluster-Pipeline gewährleistet die Genauigkeit auf Isoform-Ebene.
- Optionales naszendes RNA-Modul: 5-EU-Markierung ermöglicht die Identifizierung von naszenten Transkripten, die Schätzung der Halbwertszeit und Stabilitätsanalysen.
Integrierter Analyseplan
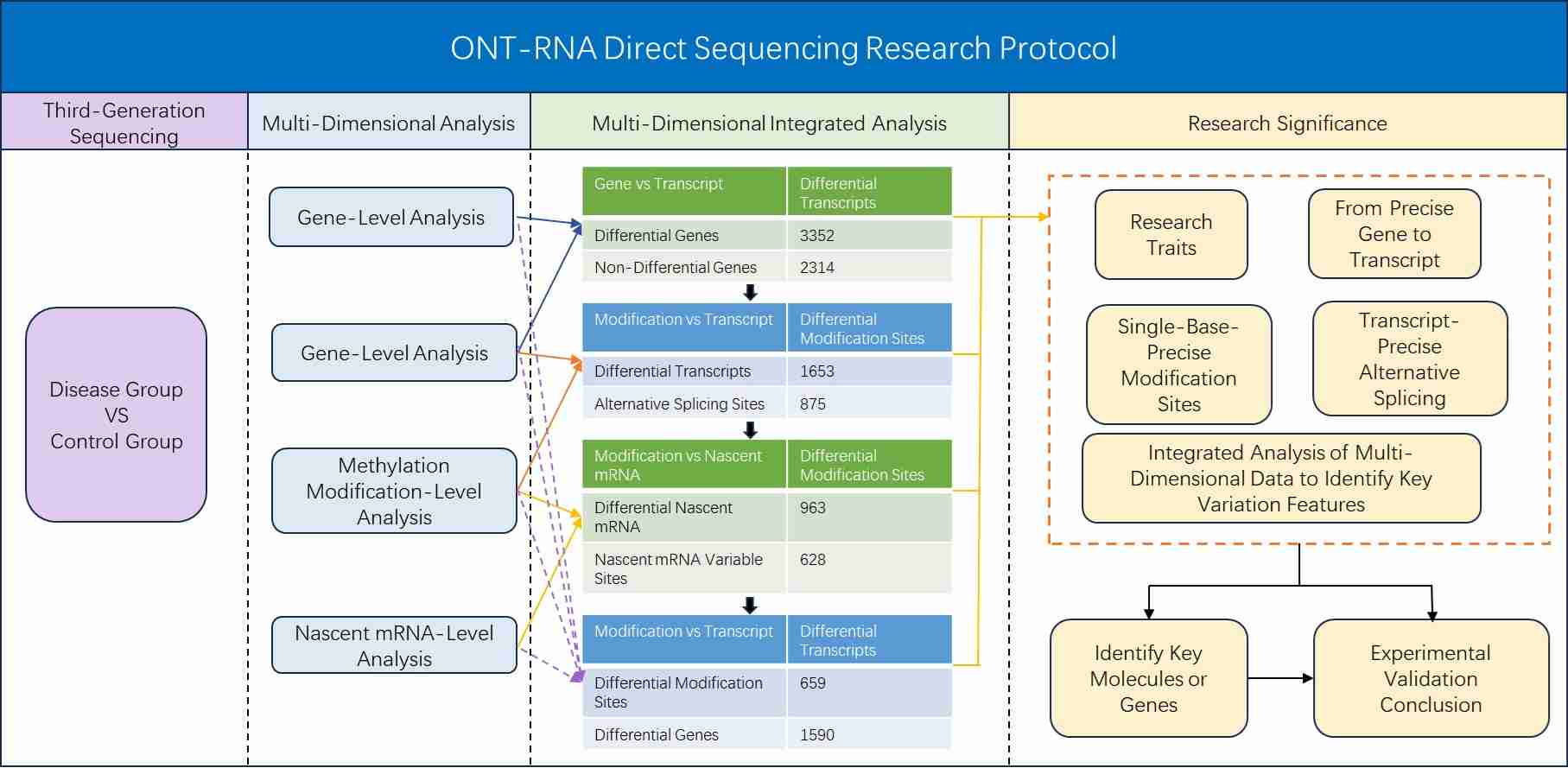
Nanopore-Direkt-RNA-Sequenzierung Bioinformatikanalyse
I. Isoform-Analyse
- Analyse des alternativen Spleißens
- Identifizierung von Fusionstranskripten
- Poly(A)-Schwanzanalyse
II. Ausdrucksquantifizierung
- Transkriptquantifizierung
- Differenzielle Transkriptanalyse
- Funktionelle Anreicherung (GO/KEGG/GSEA)
- Protein-Protein-Interaktionsnetzwerk (PPI-Netzwerk)
III. RNA-Methylierung / Modifikationsanalyse
- m6A-Stellenannotation
- Modifikationsanreicherungsanalyse
- Differenzielle m6A-Analyse
IV. Gemeinsame quantitative Expressionsanalyse der Isoformen
- AltTP-Analyse
- Funktionale Diversitätsanalyse (FDA)
- Differenzielle Anreicherungsanalyse
V. Analyse von naszente mRNA (optional)
- Identifizierung von naszente mRNA
- Halbwertszeitabschätzung
- Analyse der Korrelation der mRNA-Stabilität
- Differenzielle neuartige Expression
Was Sie von unserem Nanopore-Direkt-RNA-Sequenzierungsdienst erhalten werden
Umfassende direkte RNA-Ergebnisse
Vollständig verarbeitete Datensätze einschließlich FAST5/FASTQ, gespleißte BAM/CRAM, GTF/GFF (neue/bekannte Isoformen, TSS/TTS, poly(A)-Stellen), poly(A)-Schwanz-Tabellen, Listen von RNA-Modifikationsstellen (m6A/m5C/Ψ/I), Fusionsaufrufe und Transkript-Ebenen-Zählungen/TPM—bereit für die sofortige Analyse mit Nanopore-Direkt-RNA-Sequenzierung und nachgelagerte Verwendung.
Differenzielle und funktionale Einblicke
Bedingungsspezifische Ergebnisse, die differenzielle Transkripte, AltTP/DIU, differenzielles m6A und poly(A)-Vergleiche umfassen, sowie GO/KEGG-Anreicherung und optionale PPI-Netzwerküberlagerungen, um Isoform-/Modifikationsänderungen mit Signalwegen zu verknüpfen.
Veröffentlichungsbereite Visualisierungen
Hochwertige Abbildungen: Isoform-Hitzekarten, Vulkanplots, Sashimi/AltTP/DIU-Visualisierungen, Fusion-Breakpoint-Panels, Modifikations-Metagene/Motivansichten und poly(A)-Histogramme – optimiert für die Interpretation und die Einreichung bei Fachzeitschriften.
Transparente Analyseaufzeichnungen
Ein prägnanter Methoden- und QC-Bericht (Zusammenfassung des Direct RNA 3′→5′ Protokolls, Software-Versionen/Parameter, Ertrag/Lesequalität/Mapping/Junction-Metriken) mit Pipeline-Manifest, Umgebungsdatei und Prüfziffern für vollständige Reproduzierbarkeit.
Optionale Zusatzleistungen
Entstehendes RNA-Modul (5-EU-Identifikation, Halbwertszeit, Stabilitätskorrelationen), integrative Multi-Omik (z.B. Illumina-Tiefe + direkte RNA-Merkmale), Nicht-Poly(A)-Erfassungsstrategien, interaktive HTML-Dashboards und benutzerdefinierte Exporte (z.B. Cytoscape-bereit).
Musteranforderungen & Versand
| Kategorie | Anforderungen & Hinweise |
|---|---|
| Standard-Eingabe | Gesamt-RNAempfohlen ≥50 µg bei ≥180 ng/µL; brauchbar 20–80 µg (zielabhängig). ZellführerEtwa 1×10^7 Zellen liefern typischerweise ausreichend RNA. RNA-TypPoly(A)+ mRNA standardmäßig; nicht-poly(A) Optionen verfügbar (benutzerdefinierte Erfassung/NERD-ähnlich). |
| Qualitätskriterien | Integrität: RIN ≥7 oder hohe DV200. SauberkeitVermeiden Sie Phenol/EDTA-Rückstände und DNase-Inhibitoren. |
| Replikate & Kontrollen | Replikate≥2 biologische Proben pro Bedingung (≥3 bevorzugt für differentielles/AltTP). Steuerelemente (optional)ERCC-Spike-Ins, Methylierungsstandards (zur Kalibrierung des Modifikationsaufrufs). |
| Einreichung & Versand | VerpackungRNase-freie Röhrchen, deutlich beschriftet (Projekt-ID, Proben-ID, Konzentration, Volumen). Temperatur: Schiff auf Trockeneis. Manifest: Beispielmetadaten einfügen (Organismus, Behandlung, beabsichtigte Vergleiche). EinhaltungBefolgen Sie internationale Vorschriften für Biomaterialien; fügen Sie das Sicherheitsdatenblatt (SDS) hinzu, falls erforderlich. |
| Niedrigeingabe / Sonderfälle | Konsultieren Sie uns für RNA-Strategien mit geringem Input, gezielte Erfassungs-Panels (lncRNA/circRNA) oder degradierte/archivierte Gewebe (Machbarkeit von Fall zu Fall). |
Anwendungen & Fallstudien
Kernanwendungen
- Isoform-resolvente Transkriptomik: Voll-Längen-Isoformen, alternatives Spleißen (AltTP/DIU), TSS/TTS und Nutzung von Poly(A)-Stellen.
- Fusion-Transkript-ErkennungEinzelmolekülbeweise über Bruchstellen für hochkonfidente Aufrufe.
- EpitranskriptomikTranskriptom-weite RNA-Modifikationskartierung (m6A/m5C/Ψ/I) mit differentieller Analyse.
- Poly(A)-Schwanzbiologie: vor dem Lesen gemessene Poly(A)-Schwanzlängen zur Untersuchung von Stabilität und Translation; isoformspezifische Unterschiede.
- Entstehende RNA & Zerfall (optional)5-EU-basiertes naszentes RNA, Halbwertszeitschätzung, Stabilitätskorrelationen.
- Nicht-kodierende RNAEntdeckung/Quantifizierung von lncRNA und circRNA mit langen Reads.
- Genomannotation: neuartige Transkripte/Gene in Nicht-Modellorganismen oder heterogenen Geweben.
- Längsdynamikgleichzeitige Veränderungen in Struktur, Ausdruck und Modifikationen über Zeit oder Bedingungen hinweg.
Fallübersichten
- Viral-RNABasislevel-m6A-Kartierung auf vollständigen Genomen, um Modifikationen mit Spleißung/Isoformen in Beziehung zu setzen.
- Hepatitis-Virus-ModelleDirekte Detektion von m5C auf viralen RNAs zur Erforschung von Export- und angeborenen Erkennungspfaden.
- OnkologiePseudouridin (Ψ) Dynamik im Zusammenhang mit der Kontrolle der Translation; Fusionstranskripte in Tumorproben.
- Pflanzen-/LichtreaktionenVollständige Lesevorgänge zeigen lichtregulierte posttranskriptionale Spleißereignisse.
- Stoffwechselzuständegewebespezifische poly(A)-Änderungen und Isoformenwechsel unter Fasten/Essen.
- StressphysiologieLangzeitblutprofilierung zeigt gleichzeitige Veränderungen in der Expression und m6A-Markierungen.
Methodenvergleich
| Merkmal | Illumina KurzleseRNA-Seq | ONT cDNA Langzeitlesung (RT/PCR) | ONT Direkte RNA (Dieser Dienst) |
|---|---|---|---|
| Molekülauslesung | cDNA-Fragmente | cDNA Volllängen (amplifiziert) | Native RNA, Einzelmolekül |
| Vollständige Isoformen | Impliziert/mehrdeutig | Ja | Ja (keine Montage) |
| RT/PCR-Bias und GC-Effekte | Geschenk | Präsent (reduziert vs. SR) | Minimal (PCR-frei) |
| RNA-Modifikationen | Indirekt/prüfungs-spezifisch | Verloren während RT | Direkte m6A/m5C/Ψ/I-Signale |
| Poly(A)-Schwanzlänge | Nicht zugänglich | Indirekt/nicht pro Lesen | Vorab-Leselängen |
| Fusion-Transkript-Detektion | Herausfordernd (Montage) | Gut | Hohe Zuversicht (einzelner Lesevorgang über den Bruchpunkt) |
| Quantifizierungsgenauigkeit | Betroffen von Fragmentierung/Modellierung | RT/PCR-Bias möglich | Einheimisch, strand-spezifisch, reduzierte Verzerrung |
| Durchsatz | Höchste | Moderat | Niedriger vs cDNA (höhere Detailgenauigkeit pro Molekül) |
| Am besten für | Sehr große Kohorten; Genebene DE | Isoformen in hoher Ausbeute (keine Modifikationen) | Isoformen + Modifikationen + poly(A) auf denselben Molekülen |
| Typische Verwendung | Expressions-Screening, Bulk-DE | Isoform-Kataloge, lange UTRs | Mechanismusfokussierte Studien: Spleißen/AltTP, Epitranskriptom, Stabilität |
Warum CD Genomics
1. Native-RNA-Expertise
Direkte RNA 1D, PCR-frei, 3′→5′ Orientierung; validiertes Direct RNA Sequencing Nanopore-Protokoll mit strengen Proben-/Lauf-QC.
2. End-to-End-Workflow & Analyse
Von der Proben-QC und Bibliotheksvorbereitung bis hin zur Sequenzierung, Nanopore-Direkt-RNA-Sequenzierungsanalyse und integrierter Berichterstattung—Isoformen, m6A/m5C/Ψ/I, poly(A)-Schwanz.sund optionale naszierende RNA.
3. In peer-reviewed Forschung nachgewiesen
Nanopore-Direkt-RNA-Bibliotheken, die von CD Genomics vorbereitet wurden, wurden in einer PLOS Genetics-Studie über Arabidopsis Epitranskriptomik, die Einzelmolekül-m6A-Kartierung mit DRS demonstrierend (Sun et al., 2022).
4. Qualität & Reproduzierbarkeit
Transparente Methoden & QC-Bericht, versionsgesperrte Pipelines, Manifeste und Prüfziffern; veröffentlichungsbereite Abbildungen und wiederverwendbare Datenpakete.
5. Entwickelt für Ihre Frage
Studienentwurf für Isoformen/Spleißung, Epitranskriptom, Poly(A)-Biologie, naszente RNA; Optionen für die Nicht-Poly(A)-Erfassung und Multi-Plattform-Integration.
Demonstrationsergebnisse
Teilweise Ergebnisse sind unten aufgeführt:
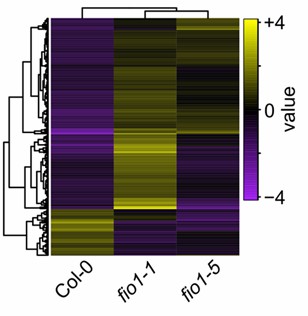
Differenzielle Isoform-Expressions-Wärmekarte
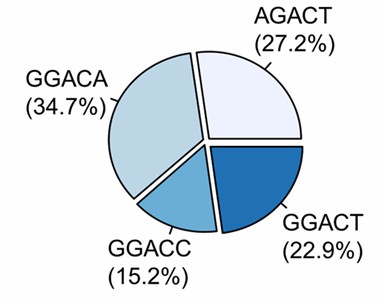
Verteilung der m6A-Stellen über Transkriptregionen
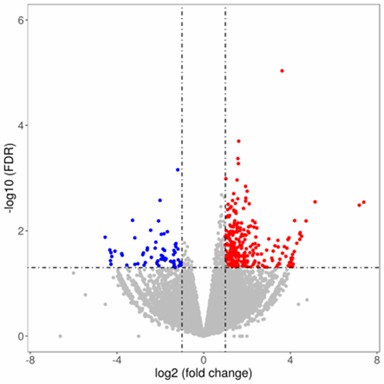
Differentiale Isoform-Volcano-Diagramm
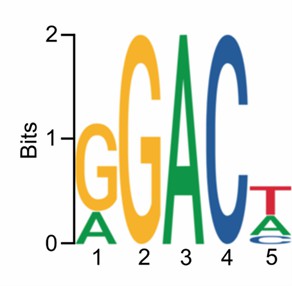
m6A Sequenzmotiv-Logo
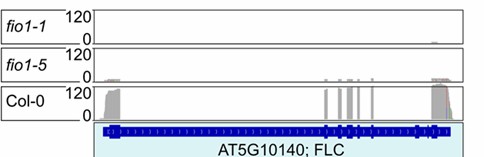
Sequenzabdeckung an einem Genlocus
Referenz:
- Sonne, Bin, et al. "Die FIONA1-vermittelte Methylierung des 3'UTR von FLC beeinflusst die FLC-Transkriptspiegel und die Blüte in Arabidopsis.." PLoS Genetik 18.9 (2022): e1010386.
Nanopore Direct RNA-Sequenzierung FAQs
Q1. Wann ist Direct RNA nicht die erste Wahl für die Analyse?
Für große, genebasierte Differenzialexpressionsscreenings mit engen Budgets bietet cDNA/Illumina in der Regel mehr Tiefe pro Dollar. Der Durchsatz von Direct RNA ist geringer und die Kosten pro nutzbarem Read sind höher, daher ist es am besten, es für Fragestellungen zu reservieren, bei denen native RNA-Merkmale von Bedeutung sind – vollständige Isoformen, alternatives Splicing, Fusionstranskripte, RNA-Modifikationen und Poly(A)-Schwanzbiologie.
Q2. Wie unterscheidet sich das Nanopore Direct RNA-Sequencing von herkömmlichem RNA-seq?
Traditionelles RNA-seq wandelt RNA in cDNA um und amplifiziert es durch PCR, was Bias einführen und native chemische Markierungen entfernen kann. Nanopore Direct RNA liest native RNA-Moleküle direkt in einer 3′→5′-Orientierung ohne RT/PCR, bewahrt RNA-Modifikationen und ermöglicht die Messung der poly(A)-Schwänze pro Lesevorgang sowie vollständige Isoformen.
Q3. Was sind die Hauptprobleme der direkten RNA?
Direkte RNA hat derzeit eine niedrigere Durchsatzrate als cDNA und eine höhere Einzel-Lese-Fehlerrate als Illumina, und es erfordert spezialisierte, signalbewusste Analysen und ausreichende Rechenleistung. In der Praxis mildern wir diese Einschränkungen durch angemessenes experimentelles Design, Konsens über die Reads, robuste Ausrichtung und sorgfältige Qualitätskontrolle.
Q4. Kann Direct RNA die Analyse von Genexpression, Splicing-Isoformen und Fusions-Transkripten durchführen?
Ja, vorausgesetzt, die Studie ist für diese Endpunkte ausgelegt. Wir quantifizieren routinemäßig Transkripte, analysieren die Isoformnutzung (AltTP/DIU) und detektieren Fusionstranskripte mit Einzelmolekülbeweisen; jedoch kann cDNA/Illumina für sehr große Kohorten, die sich ausschließlich auf die genebene DE konzentrieren, wirtschaftlicher sein, während Direct RNA für mechanismusgesteuerte Studien bevorzugt wird, bei denen native RNA-Merkmale entscheidend sind.
Q5. Können Poly(A)-Schwänze und RNA-Modifikationen in demselben Durchlauf profiliert werden?
Ja. Sowohl die Länge des poly(A)-Schwanzes als auch Modifikationssignaturen wie m6A, m5C und Ψ/Inosin stammen aus demselben Nanopore-Signal auf nativer RNA, was eine gemeinsame Interpretation der Moleküle ermöglicht, die die Merkmale tragen.
Q6. Ist die direkte RNA-strangspezifisch, und wie steht es um die 5′-Trunkierung?
Reads sind von Natur aus strangspezifisch und verlaufen 3′→5′ von einem Poly(T)-Adapter. Einige Reads sind chemisch und durch die Dynamik der Pore 5′-verkürzt; unsere Schritte zur Isoform-Clusterbildung und Entredundanzierung berücksichtigen dies, sodass die Aufrufe auf Isoform-Ebene zuverlässig bleiben.
Q7. Welche Eingaben benötige ich?
Wir empfehlen ≥50 µg Gesamt-RNA bei ≥180 ng/µL, mit einem praktikablen Bereich von 20–80 µg, abhängig von Zielen und Qualität; intakte RNA (z. B. RIN ≥ 7 oder hoher DV200) verbessert die Ergebnisse erheblich. Siehe Probenanforderungen für Versand- und QC-Details.
Q8. Erfassen Nanopore Direct RNA-Lesungen m6A direkt?
Ja. Da native RNA die Pore ohne RT/PCR passiert, erzeugt m6A charakteristische Stromsignaturen, die mit nahezu basenaufer Auflösung erfasst und spezifischen Transkripten und Isoformen zugeordnet werden können.
Q9. Wie viel RNA wird für die ONT Direct RNA-Sequenzierung benötigt?
Die meisten Projekte zielen auf ≥50 µg Gesamt-RNA bei ≥180 ng/µL ab; Studien mit 20–80 µg können je nach Zielen und RNA-Integrität machbar sein, was die Ausbeute und die Analyse-Tiefe stark beeinflusst.
Q10. Wie unterscheidet sich Direct RNA von cDNA-Langlese- oder Illumina-Kurzlese-RNA-Seq?
Direkte RNA-Lesungen erfassen natives RNA (3′→5′) ohne RT/PCR, bewahren RNA-Modifikationen und ermöglichen poly(A) pro Lesung. cDNA/Short-Reads verlieren native Modifikationen und leiten Isoformen aus Fragmenten ab.
Nanopore Direkte RNA-Sequenzierung Fallstudien
Kundenveröffentlichungshighlight
Journal: PLoS Genetik
AutorSun, B., Bhati, K. K., Song, P., et al.
Veröffentlicht27. September 2022
1) Hintergrund
Blütezeit in Arabidopsis thaliana wird streng durch RNA-regulatorische Schichten kontrolliert, einschließlich N6-Methyladenosin (m6A). Sonne u. a.untersuchte, ob der m6A-Schreiber FIONA1 (FIO1; METTL16-ähnlich) Methylmarkierungen an der 3′UTR von FLC und stabilisiert damit das Transkript. Um eine unverzerrte, einzelmolekulare Sicht auf RNA-Methylierung und Isoformabdeckung zu erhalten, integrierte das Team Illumina RNA-Seq, MeRIP-seqund Nanopore Direct RNA-Sequenzierung (DRS).
2) Methoden
- 14-Tage Arabidopsis thaliana Setzlinge (WT Col-0; fio1-1, fio1-5).
- Bibliotheksvorbereitung: ONT SQK-RNA002 Direkt-RNA-Protokoll; Bibliotheken vorbereitet von CD Genomics.
- Sequenzierung: PromethION (R9.4), ~48–72 h Läufe.
- Basecalling/QC: Guppy v3.2.6; Filterung mit NanoFilt.
- Ausrichtung & Korrektur: minimap2-Mapping; Fclmr2-Korrektur; Metriken über samtools.
- m6A-Analyse & Statistiken: Tombo (de novo) + MINES; differentielle Methylierung mit methylKit.
3) Ergebnisse
Die Analyse der direkten RNA-Sequenzierung mit Nanoporen offenbarte genotypabhängige Transkriptomveränderungen und ein m6A-Konsens von RGACH in Arabidopsis. Im Wildtyp traten die meisten m6A-Ereignisse bei GGACA (34,7 %) auf, gefolgt von AGACT (27,2 %), GGACT (22,9 %) und GGACC (15,2 %). Über fio1 Mutanten, DRS identifizierte 74 hypomethylierte Gene (fio1-1) und 63 (fio1-5) im Vergleich zu WT. Kritisch ist, dass die DRS-Abdeckung die Depletion von bestätigte. FLC mRNA in fio1-1 und fio1-5, konsistent mit dem Verlust der Methylierung des 3′UTR.
4) Schlussfolgerungen
Diese Studie zeigt, wie die Nanopore Direct RNA Sequencing eindeutige Nachweise auf Einzelmolekülebasis liefert, die 3′UTR m6A mit der Stabilität von mRNA verknüpfen. Durch die Erhaltung von nativer RNA koppelt DRS direkt die Erkennung von m6A-Stellen, die Entdeckung von Motiven und die Abdeckung auf Isoform-Ebene – und stellt fest, dass FIO1 m6A an der FLC 3′UTR, und der Verlust dieses Merkmals führt zu einer schnellen FLC Zersetzung und frühe Blüte.
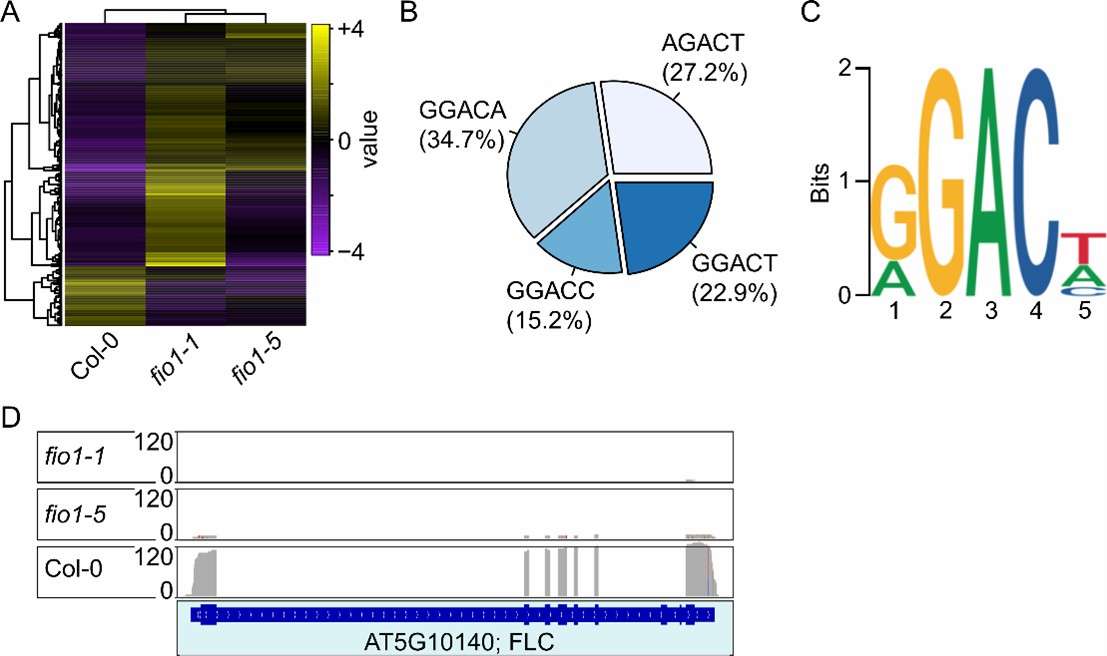 Direkte RNA-Sequenzierungsanalyse.
Direkte RNA-Sequenzierungsanalyse.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Die von FIONA1 vermittelte Methylierung der 3'UTR von FLC beeinflusst die FLC-Transkriptspiegel und die Blüte in Arabidopsis.
Journal: PLoS Genetik
Jahr: 2022
Der m6A-Schreiber FIONA1 methylisiert die 3'UTR von FLC und steuert die Blüte in Arabidopsis.
Journal: bioRxiv
Jahr: 2022
Vollständige Genomsequenz des lignocellulose-abbauenden Actinomyceten Streptomyces albus CAS922
Journal: Mikrobiologie Ressourcenankündigungen
Jahr: 2020
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


