
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Menschliche mitochondriale DNA (mtDNA) Sequenzierung
Einführung in die Sequenzierung der menschlichen mitochondrialen DNA (mtDNA)
Mitochondrien, die als "Kraftwerke der Zelle" bezeichnet werden, sind lebenswichtige Organellen in menschlichen Zellen. Die menschliche mitochondriale DNA (mtDNA) bildet doppelt-strängige zirkuläre Moleküle, die etwa 16.569 DNA-Basenpaare enthalten und 37 Gene kodieren (Abb. 1). Es gibt Tausende von mtDNA-Molekülen, und mtDNA ist hoch variabel, da sie nicht durch Histone geschützt ist. Die mutierte mtDNA koexistiert mit den Wildtyp-Molekülen in einem Zustand, der als Heteroplasmie bezeichnet wird, und wurde mit einer Vielzahl von menschlichen Krankheiten in Verbindung gebracht, wie Autismus-Spektrum-Störung (ASD), Lebersche hereditäre Optikusneuropathie (LHON), Brustkrebs, Alzheimer-Krankheit, Parkinson-Krankheit und Bluthochdruck. Daher ist das mitochondriale Genom ein bedeutendes Forschungsobjekt in der klinischen Diagnostik.
CD Genomics hat Panels entwickelt, die das gesamte menschliche mitochondriale DNA-Genom spezifisch erfassen. Diese basieren auf multiplex PCR und Probenfang. gezielte Sequenzierung Ansätze, die das menschliche mitochondriale Genom zu 100 % abdecken könnten. Mit diesem Panel müssen Kunden kein gereinigtes mtDNA mehr bereitstellen. Die Anreicherung von mtDNA erfolgt durch Multiplex-PCR-Amplifikation oder Probenfang des mitochondrialen Genoms. Die angereicherte mtDNA wird dann einer Bibliotheksvorbereitung und ultra-tiefen Sequenzierung unterzogen. CD Genomics bietet diese Technologie an, um Kunden bei der Forschung zu mitochondrialen genombezogenen Krankheiten zu unterstützen.
 Abbildung 1. Das zirkuläre menschliche mitochondriale Genom.
Abbildung 1. Das zirkuläre menschliche mitochondriale Genom.
Vorteile unseres Sequenzierungsdienstes für menschliche mitochondriale DNA (mtDNA)
- Einfacherer Zugang zu Ausgangsproben: menschliche gDNA, die sowohl mtDNA als auch nukleare DNA enthält.
- Ultra-tiefe Sequenzierung: >1000x Abdeckung, mit der Illumina PE150 Plattform
- Hohe Genauigkeit: 100 % Amplikon- oder Sondenabdeckung aller Regionen des mitochondrialen Genoms
- Niedrigster Preis: abhängig von der Stichprobengröße, bitte kontaktieren Sie uns für ein Angebot.
- Schnellste Bearbeitungszeit: 3-4 Wochen
Workflow zur Sequenzierung der menschlichen mitochondrialen DNA (mtDNA)

Dienstspezifikationen
Beispielanforderungen
|
|
 Klicken |
Sequenzierungsstrategien
|
 |
Bioinformatikanalyse Der Standard-Service für die Bioinformatik von mtDNA-Sequenzierungen des Menschen umfasst
|
Analyse-Pipeline
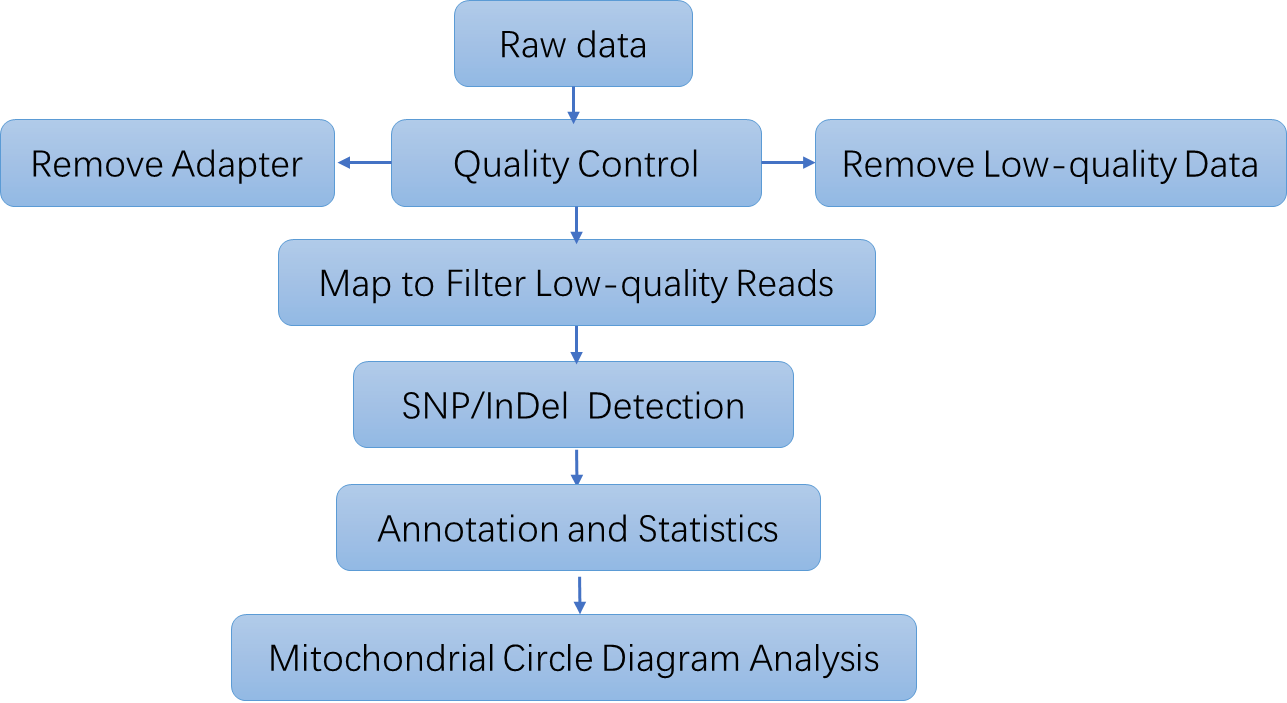
Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details zur Sequenzierung der menschlichen mitochondrialen DNA (mtDNA) für Ihre Schreibanpassung.
Demonstrationsergebnisse
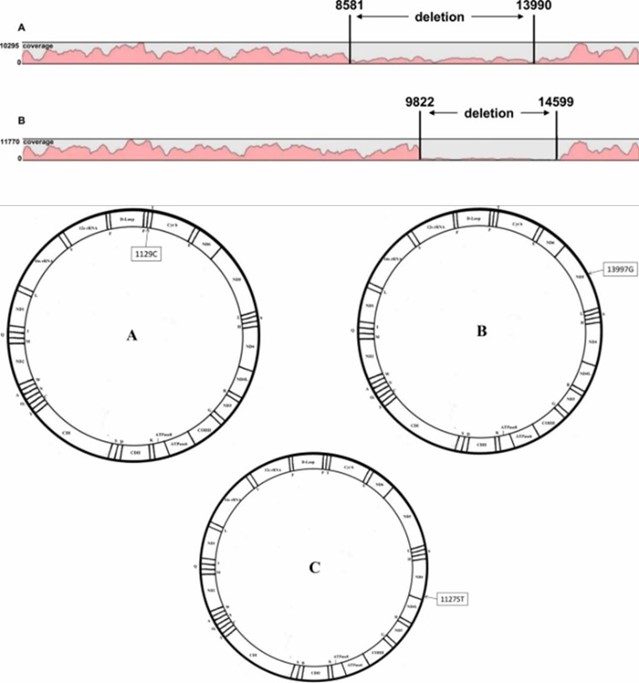 Kartierung und Abdeckungsergebnisse der mtDNA-Sequenzierung sowie die Darstellung von mtDNA-pathologischen Profilen. (Yao et al., 2019)
Kartierung und Abdeckungsergebnisse der mtDNA-Sequenzierung sowie die Darstellung von mtDNA-pathologischen Profilen. (Yao et al., 2019)
Referenz:
- Yao Y, Nishimura M, Murayama K, et al. Eine einfache Methode zur Sequenzierung des gesamten menschlichen mitochondrialen Genoms direkt aus Proben und deren Anwendung in der genetischen Testung. Wissenschaftliche Berichte, 2019, 9(1): 17411.
Menschliche mitochondriale DNA (mtDNA) SeqFAQs
1. Beinhaltet die Ganzgenomsequenzierung mitochondriale DNA?
Whole-Genome-Sequenzierung (WGS) umfasst von Natur aus mitochondrialer DNA in seinem Umfang. Diese umfassende Sequenzierungstechnik beinhaltet die Extraktion und Sequenzierung sowohl von nukleärer als auch von mitochondrialer DNA. Aufgrund der Fülle an mtDNA-Kopien innerhalb der Zellen, Whole-Genome-Sequenzierung sichert eine umfassende Abdeckung des mitochondrialen Genoms und ermöglicht eine gleichzeitige Analyse mit dem nukleären Genom.
2. Welche Methoden gibt es zur Sequenzierung von menschlicher mitochondrialer DNA?
Verschiedene Methoden werden verwendet, um die mitochondriale DNA (mtDNA) des Menschen zu sequenzieren. Zu den gängigen Ansätzen gehören Sanger-Sequenzierung, Next-Generation-Sequenzierung Plattformen (z. B. Illumina-Sequenzierung) und Langread-Sequenzierungstechnologien (z. B. Oxford Nanopore Technologien und Pacific BiosciencesJede Methodologie bietet unterschiedliche Vorteile, Nachteile und Anwendbarkeit, sodass Forscher ihre Auswahl basierend auf spezifischen Forschungszielen und Ressourcenüberlegungen anpassen können.
Fallstudien zur Sequenzierung der menschlichen mitochondrialen DNA (mtDNA)
Ein Verfahren zur multiplexen Voll-Längen-Einzelmolekül-Sequenzierung des menschlichen mitochondrialen Genoms
Zeitschrift: Nature Communications
Impactfaktor: 16,7
Veröffentlicht: 06. Oktober 2022
Hintergrund
Mitochondrien enthalten zirkuläre Genome mit vestigialen Funktionen. Heteroplasmie kann die Gesundheit erheblich beeinflussen, was eine genaue Variantenerkennung für Diagnose und Behandlung erforderlich macht. Traditionelle Sequierungsmethoden haben Einschränkungen, aber Langlesetechnologien wie ONT und PacBio bieten verbesserte Genauigkeit. Die Verwendung von Cas9 für gezielte Anreicherung mit der Q20+-Chemie von ONT verbessert die Lesegenauigkeit und senkt die Kosten, was sie für Proben von geringer Qualität geeignet macht und ihren Einsatz in der mitochondrialen Forschung und klinischen Anwendungen erweitert.
Methoden
- Humanzelllinien
- 15 klinische Proben
- DNA-Qualitätskontrolle
- Langstrecken-PCR
- Zirkuläre mtDNA-Voranreicherung
- Bibliotheksvorbereitung
- Langtext Nanoporen-Sequenzierung
- Kurzzeit-Sequenzierung
- Vorverarbeitung von Rohdaten
- Ausrichtung
- Variantaufruf
- Annotation von mtDNA-Varianten
- Circos-Diagramme
Ergebnisse
Die Autoren verfeinerten das Cas9-mtDNA-Anreicherungsprotokoll, um eine hochselektive Einzelmolekül-Sequenzierung von mtDNA aus menschlichen gDNA-Proben zu ermöglichen, was Multiplexing ohne zusätzliche Barcode-Schritte erleichtert. Diese Methode umfasst die Verdauung mit Exonuklease V zur Anreicherung von mtDNA, die Dephosphorylierung der gDNA-Enden und die sequenzspezifische Spaltung durch die dual-RNA-gesteuerte Cas9-Endonuklease. Dieser Ansatz erzeugt einen Barcode an der Cas9-Schnittstelle, was die Bibliotheksvorbereitung und Sequenzierung erleichtert. Nach der Cas9-Spaltung werden die Proben mit Proteinase K behandelt, um das gebundene Cas9-Protein zu entfernen, gefolgt von der Ligation von ONT-Sequenzierungsadaptern an die phosphorylierten Schnittstellen.
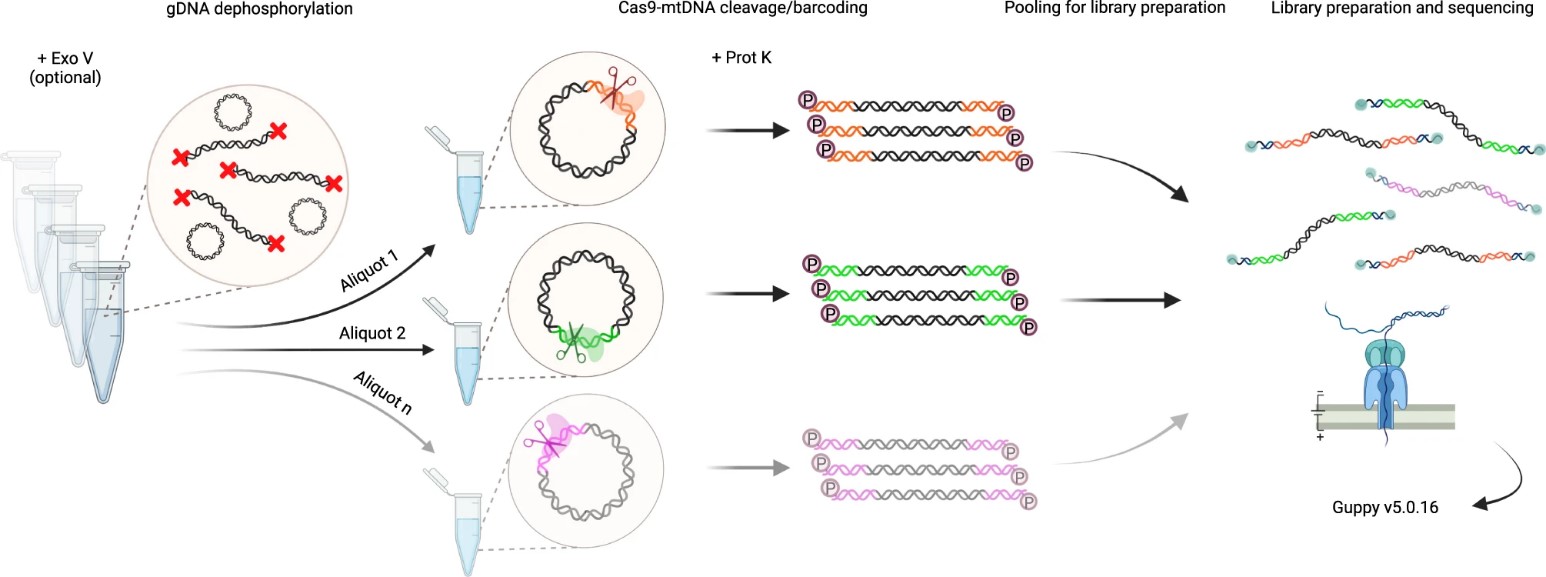 Abb. 1 Cas9-mtDNA-Anreicherung, Barcoding, Pooling und Demultiplexing-Ansatz für Langsequenzierung.
Abb. 1 Cas9-mtDNA-Anreicherung, Barcoding, Pooling und Demultiplexing-Ansatz für Langsequenzierung.
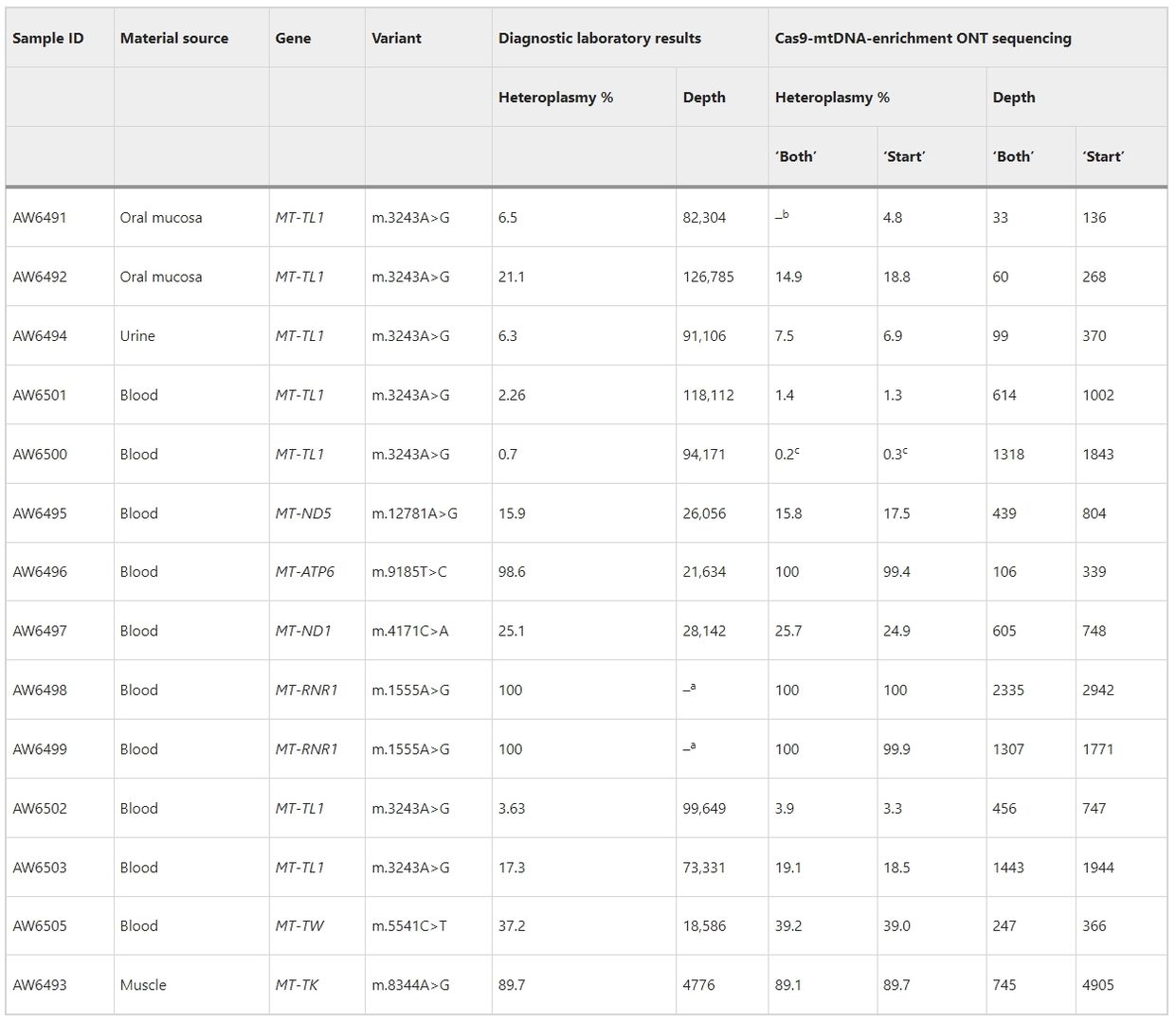
Die Cas9-mtDNA-Anreicherungsanalyse erkennt genau Heteroplasmie in pathogenen SNVs über 14 klinische Proben. Die Methode, ohne Exonuklease-V-Behandlung, erreicht eine variable mtDNA-Genomabdeckung (×33 bis ×2335), die mit den DNA-Abbaustufen der Proben korreliert. Die Einbeziehung sowohl vollständiger Reads als auch Reads, die an spezifischen Cas9-Schnittstellen-Barcodes beginnen, erhöht die Abdeckung und das Vertrauen in die Variantenbestimmung. Alle berichteten Heteroplasmien sind bestätigt, mit Frequenzen von <0,2% bis 100%, und es wurden keine großen Deletionen beobachtet. Darüber hinaus werden nicht-pathogene polymorphe Stellen und Varianten unbekannter Signifikanz identifiziert, was die Bedeutung der Bewertung der Variantenfrequenz innerhalb der allgemeinen Bevölkerung und spezifischer Haplogruppen unterstreicht.
Tabelle 1. Identifizierung pathogener SNVs und Bestimmung der Heteroplasmie in den bestätigten klinischen Proben mit mtDNA-Veränderungen
Die Cas9-mtDNA-Anreicherungsanalyse identifiziert erfolgreich mehrere mtDNA-Deletionen in einem komplexen klinischen Fall. Mit drei gRNAs, die das mitochondriale Genom abdecken, erkennt die Methode zwei große Deletionen: m.3264_16070del (7,6% Heteroplasmie) und m.10751_14129del (84,1% Heteroplasmie), neben wildtypischem mtDNA (8,3% Häufigkeit). Der Vergleich mit lrPCR und Illumina-Sequenzierung bestätigt den Vorteil der nativen mtDNA-Sequenzierung, da sie Verzerrungen überwindet und genauere Schätzungen der mtDNA-Populationen liefert.
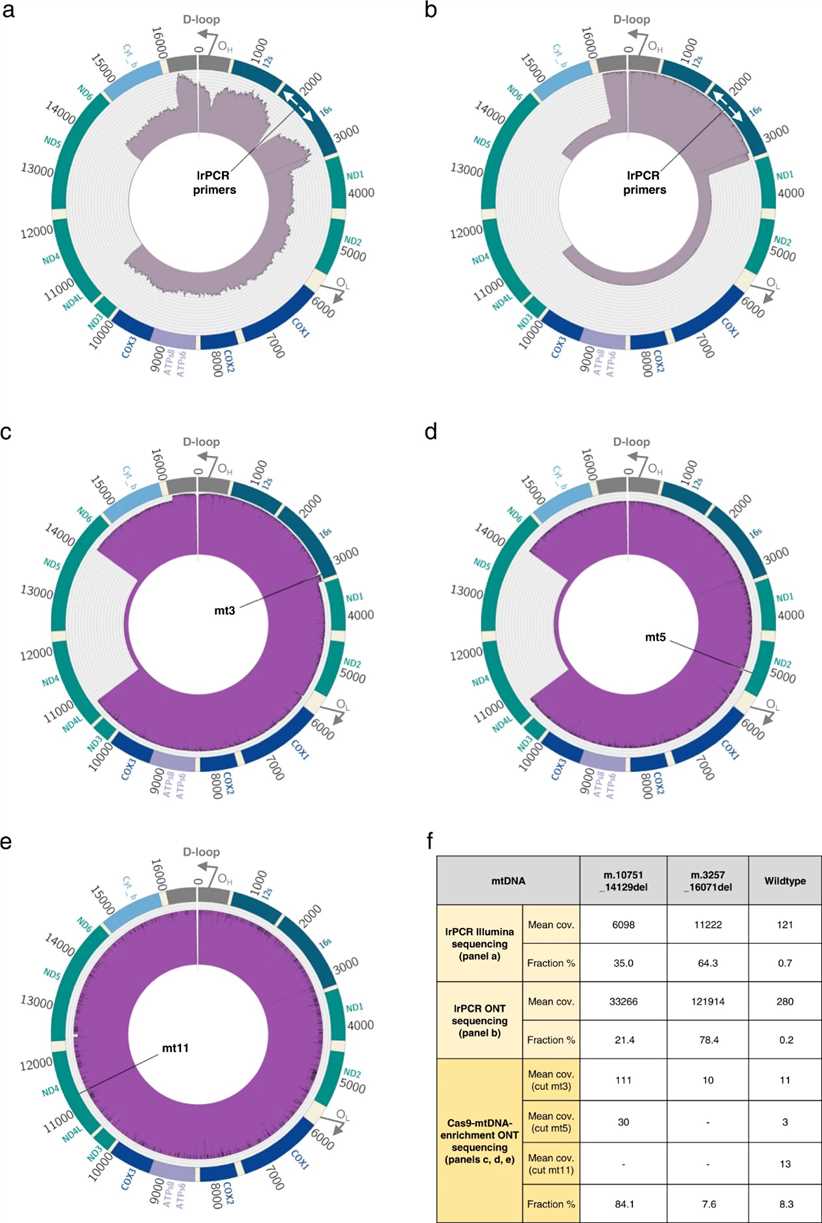 Abb. 2 Mehrfache mtDNA-Deletionen in einer klinischen Probe.
Abb. 2 Mehrfache mtDNA-Deletionen in einer klinischen Probe.
Fazit
Dieses Verfahren verwendet die Cas9-Nuklease und Nanoporen-Sequenzierung, um das vollständige menschliche mitochondriale Genom effizient zu zielen und zu sequenzieren. Es erreicht eine hohe Abdeckung und erkennt genau SNVs und multiple Deletionen in klinischen Proben. Die benutzerdefinierte Analyse-Pipeline der Autoren ermöglicht eine präzise Variantenbestimmung und erleichtert eine umfassende mtDNA-Analyse, die der molekularen Diagnostik und der Populationsforschung zugutekommt. Darüber hinaus hat es potenzielle Anwendungen bei der Untersuchung somatischer Mutationen und DNA-Modifikationen und erstreckt sich auf Tierarten für die Erhaltungsgenetik und phylogenetische Forschung.
Referenz:
- Keraite I, Becker P, Canevazzi D, et al. Eine Methode zur multiplexen Voll-Längen-Einzelmolekül-Sequenzierung des menschlichen mitochondrialen Genoms. Naturwissenschaftliche Kommunikation, 2022, 13(1): 5902.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Unterschiedliche Funktionen des Wildtyp- und R273H-Mutanten Δ133p53α regulieren unterschiedlich die Aggressivität von Glioblastomen und die durch Therapie induzierte Seneszenz.
Zeitschrift: Zellsterben & Krankheit
Jahr: 2024
Hochdichte-Kartierung und Kandidatengenanalyse von Pl18 und Pl20 in Sonnenblumen durch Whole-Genome-Resequenzierung
Zeitschrift: Internationale Zeitschrift für Molekulare Wissenschaften
Jahr: 2020
Identifizierung von Faktoren, die für die m6A mRNA-Methylierung in Arabidopsis erforderlich sind, zeigt eine Rolle für die konservierte E3-Ubiquitin-Ligase HAKAI.
Zeitschrift: New Phytologist
Jahr: 2017
Generierung eines hoch attenuierten Stammes von Pseudomonas aeruginosa für die kommerzielle Produktion von Alginat
Zeitschrift: Mikrobielle Biotechnologie
Jahr: 2019
Kombinationen von Bakteriophagen sind wirksam gegen multiresistente Pseudomonas aeruginosa und erhöhen die Empfindlichkeit gegenüber Carbapenem-Antibiotika.
Journal: Viren
Jahr: 2024
Genom-Analyse und Replikationsstudien des afrikanischen Grünen Affen Simian Foamy Virus Serotyp 3 Stamm FV2014
Journal: Viren
Jahr: 2020
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


