
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
CircRNA-Sequenzierung
Um die jüngsten Entwicklungen in der Forschung zu zirkulärem RNA (circRNA) zu unterstützen, bietet CD Genomics einen circRNA-Sequenzierungsdienst an, der die neuesten Illumina-Plattformen für eine schnelle, kostengünstige und genaue Charakterisierung nutzt.
Die Einführung der CircRNA-Sequenzierung
CircRNA ist ein neuartiges Mitglied der nicht-kodierenden RNAs (ncRNAs), das durch nicht-sequenzielle Rücksplicing von Exons, Introns oder beidem erzeugt wird. Sie zeichnen sich durch eine kovalent geschlossene Schleifenstruktur aus, wodurch sie keine 5'-Endkappen und 3'-Poly-A-Schwänze besitzen. CircRNAs sind aufgrund ihrer Nuklease-Resistenz eine hochstabile Form von ncRNAs. Als sie in den 1970er Jahren erstmals identifiziert wurden, wurden circRNAs als virale Genome oder Nebenprodukte von Fehlsplicing-Ereignissen angesehen. Neuere Forschungen haben jedoch gezeigt, dass circRNAs evolutionär über Pflanzen, Tiere und Menschen hinweg konserviert sind und dass circRNAs wichtige biologische Funktionen haben.
CircRNAs können als miRNA-Schwämme wirken und eine regulatorische Funktion in der Genexpression erfüllen. Bemerkenswerterweise sind eine Vielzahl von circRNAs in spezifischen Krankheitskontexten abnormal exprimiert, was auf ihre Assoziation mit dem Auftreten und der Entwicklung menschlicher Krankheiten hinweist. CircRNAs erfüllen auch mehrere Funktionen in zellulären Prozessen, einschließlich Vorlagen für die Translation, Regulation des alternativen Spleißens und der Genexpression, Gerüste für die Assemblierung von Proteinkomplexen und Modulatoren der rRNA- und tRNA-Biogenese. Darüber hinaus können sie zur Steigerung der Immunaktivierung für antivirale therapeutische Zwecke eingesetzt werden, da sie in die Immunregulation und Virusinfektion eingreifen.
Umfassende Erkennung von circRNAs aus Hochdurchsatz-Transkriptomdaten ist ein erster und entscheidender Schritt, um ihre Biogenese und Funktion zu untersuchen. Die Hochdurchsatz-Sequenzierung Die Kombination von rRNA/linear RNA-depletiertem RNA mit computergestützten Werkzeugen hat zur Identifizierung von Tausenden neuen circRNAs und zur quantitativen Analyse ihrer linearen Wirtstranskripte in verschiedenen Organismen geführt. Im Gegensatz zu miRNAs und anderen kleinen RNAs lassen sich circRNAs nicht leicht von anderen RNA-Spezies durch Größe oder elektroforetische Mobilität trennen. Folglich werden sie in der Regel in rRNA-depletierten Bibliotheken zurückgehalten und in mit RNase R behandelten Bibliotheken angereichert, die nur lineare RNA abbaut.
Vorteile der CircRNA-Sequenzierung
- Identifiziert bekannte und neuartige circRNAs
- Ermöglicht die Profilerstellung von circRNAs über ein breites dynamisches Spektrum.
- Erforscht neuartige Biomarker und regulatorische Netzwerke von circRNAs.
Anwendungen der CircRNA-Sequenzierung
CircRNA-Sequenzierung kann für, aber nicht beschränkt auf, die folgenden Forschungsbereiche verwendet werden:
- Untersuchung der pathogenetischen Mechanismen von Krankheiten wie Krebs;
- Untersuchung von Veränderungen in der Genexpressionsregulation während Wachstum und Entwicklung;
- Forschung zu anderen Aspekten der Gen-Transkription und der Regulierung der Genexpression.
CircRNA-Sequenzierungs-Workflow
Der allgemeine Arbeitsablauf für circRNA-Sequenzierung ist unten skizziert. Um eine circRNA-Sequenzierungsbibliothek zu erstellen, besteht der erste Schritt darin, rRNA zu depletiert, gefolgt von der Verdauung linearer RNA und der strangspezifischen Bibliotheksvorbereitung. Unser hochqualifiziertes Expertenteam führt das Qualitätsmanagement durch und überwacht jeden Schritt, um zuverlässige und unvoreingenommene Ergebnisse zu gewährleisten.

Dienstspezifikation
Musteranforderungen
|
|
 Klicken |
Sequenzierungsstrategien
|
 |
Datenanalyse Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an:
Hinweis: Die empfohlenen Datenoutputs und Analyseinhalte, die angezeigt werden, dienen nur zur Referenz. Für detaillierte Informationen bitte Kontaktieren Sie uns mit Ihren maßgeschneiderten Anfragen. |
Analyse-Pipeline
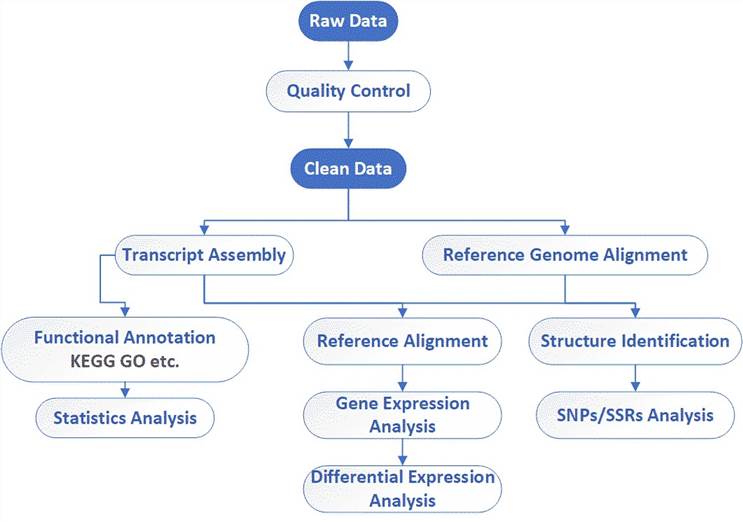
Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details zur CircRNA-Sequenzierung für Ihre Schreibanpassung.
Unterstützt von unseren erfahrenen Wissenschaftlern und fortschrittlicher Technologie kann CD Genomics Ihnen helfen, die circRNA-Sequenzinformationen mit Einzelbasenauflösung auf einmal zu erlangen durch die Hochdurchsatz-Sequenzierung durch strenge Qualitätskontrollen und fortschrittliche bioinformatische Analysen. Wenn Sie zusätzliche Anforderungen oder Fragen haben, zögern Sie bitte nicht, kontaktieren Sie uns.
Referenz:
- Wang M, Yu F, Wu W, u. a.Zirkuläre RNAs: Eine neuartige Art von nicht-kodierender RNA und ihre potenziellen Auswirkungen auf die antivirale Immunität. Internationale Zeitschrift für biologische Wissenschaften, 2017, 13(12): 1497.
Demo-Ergebnisse
Teilweise Ergebnisse sind unten aufgeführt:

Sequenzierungsqualitätsverteilung
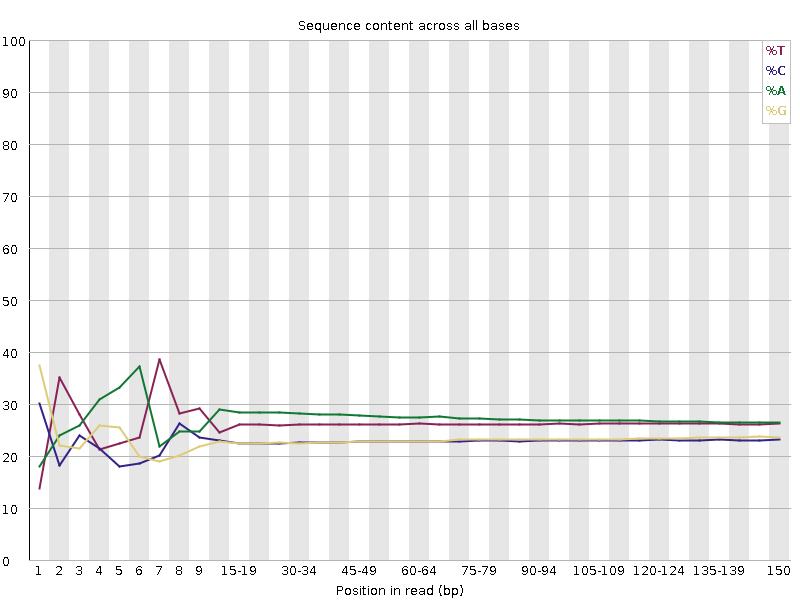
A/T/G/C-Verteilung
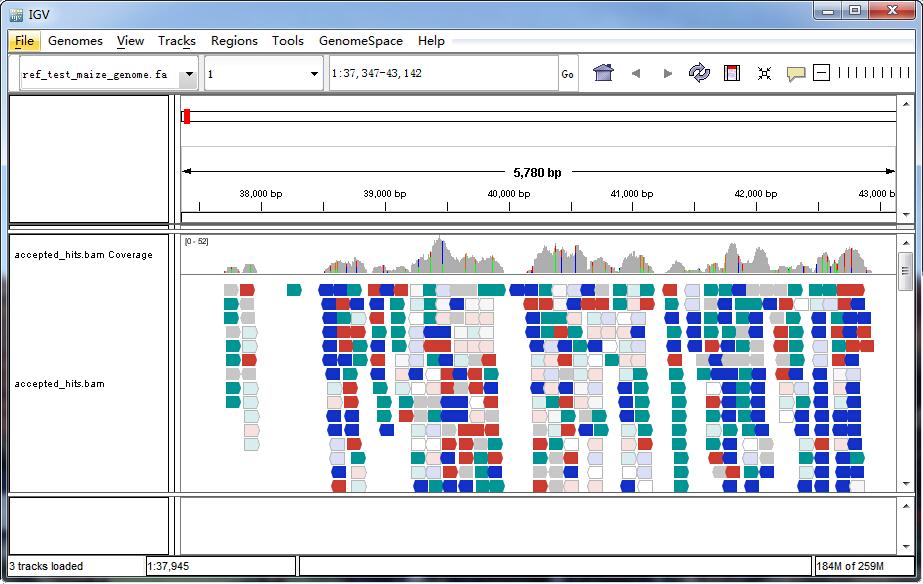
IGV-Browser-Oberfläche
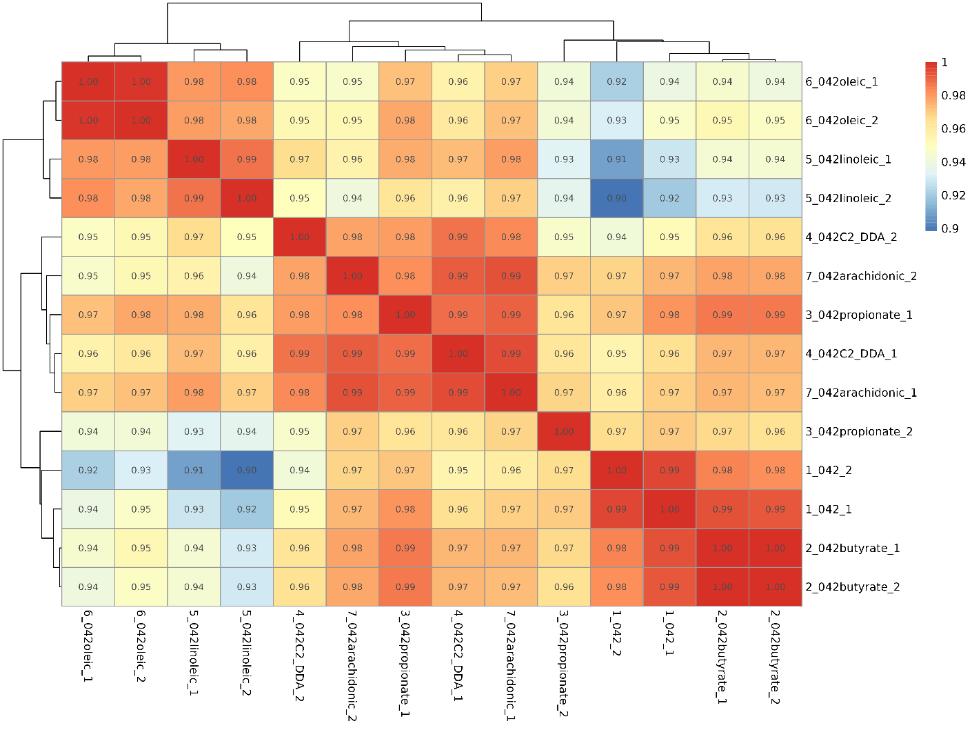
Korrelationsanalyse zwischen Proben
PCA-Score-Diagramm

Venn-Diagramm
Volcano-Diagramm
Statistik Ergebnisse der GO-Annotation
KEGG-Klassifikation
CircRNA-Seq FAQs
1. Wie wird circRNA erzeugt?
CircRNAs werden allgemein durch das Rücksplicing von Exons in nicht-sequenzieller Reihenfolge erzeugt. Die Zirkularisierung von RNA kann durch repetitive Sequenzen erfolgen, um das Rücksplicing von nicht-sequenziellen Exons zu erleichtern.
 Abbildung 1. CircRNA-Biogenese (Fischer & Leung 2016).
Abbildung 1. CircRNA-Biogenese (Fischer & Leung 2016).
2. Warum hochdurchsatztechnologie zur Analyse von circRNAs anstelle von Mikroarrays verwenden?
Hochdurchsatz-Sequenzierung ist eine zeit- und kostensparende Methode mit vielen Vorteilen gegenüber DNA. Mikroarrays.
i. Erkennung neuer Transkripte. Mikroarrays basieren auf der Hybridisierung mit spezifischen Sonden für Arten oder Transkripte, die auf bekannte circRNAs beschränkt sind. Allerdings, RNA-Sequenzierung kann neuartige Transkripte, Genfusionen, einzelne Nukleotidvarianten, kleine Einsätze und Deletionen sowie andere zuvor unbekannte Veränderungen erkennen.
ii. Breiterer dynamischer Bereich. Mikroarrays sind am unteren Ende durch den Hintergrund und am oberen Ende durch die Signalsättigung begrenzt, während die RNA-Sequenzierung diskrete, digitale Sequenzierungslesezahlen quantifizieren kann und somit einen breiteren dynamischen Bereich bietet.
iii. Erhöhte Sensitivität und Spezifität. Im Vergleich zu Mikroarrays bietet die RNA-Sequenzierung eine erhöhte Sensitivität und Spezifität, die eine verbesserte Erkennung von Genen, Transkripten und differentieller Expression ermöglicht.
iv. Nachweis von niedermolekularen und seltenen Transkripten. Die Sequenzierungstiefe kann leicht erhöht werden, um seltene Transkripte, einzelne Transkripte pro Zelle oder schwach exprimierte Gene nachzuweisen.
3. Gibt es Tipps zum Konstruieren eines circRNA-Expressionsvektors für circRNA?
Ja. Sequenzen für die Zirkularisierung und Sequenzen, die die Zirkularisierung aktivieren, sind erforderlich, um einen circRNA-Expressionsvektor zu konstruieren, der circRNAs trägt.
Referenz:
- Fischer J W, Leung A K L. CircRNAs: ein Regulator von zellulärem Stress. Kritische Bewertungen in Biochemie und Molekularbiologie, 2017, 52(2): 220-233.
CircRNA-Seq Fallstudien
CircHLA-C spielt eine wichtige Rolle bei der Lupusnephritis, indem es miR-150 bindet.
Journal: Molekulare Therapie: Nukleinsäure
Impact-Faktor: 6,392
Veröffentlicht: 12. Januar 2018
Zusammenfassung
Zirkuläre RNAs sind an der Pathogenese mehrerer Krankheiten beteiligt, indem sie als miRNAs Schwämme. Die vorherigen Studien haben berichtet, dass miR-150 positiv mit dem chronischen Nierenindex bei Patienten mit Lupusnephritis (LN) korreliert. Die Autoren untersuchten weiter das zirkuläre RNA-Profiling der Nieren und die Interaktion zwischen cirRNAs und miR-150 bei LN-Patienten. Die Bioinformatik Die Analyse sagte voraus, dass miR-150 durch circHLA-C reguliert wurde und dass circHLA-C wahrscheinlich eine wichtige Rolle in der Pathogenese von LN spielt, indem es miR-150 abfängt.
Materialien & Methoden
- Menschliche Nierengewebeproben
- Sieben LN-Patienten
- RNA-Extraktion
- Bibliothekskonstruktion
- circRNA-Sequenzierung
- HiSeq 4000 Sequenzierungssystem
- Echtzeit-qPCR
- Hierarchische Clusteranalyse
- Differenzielle Expressionsanalyse
- GO- und KEGG-Analyse
- Statistische Analyse
Ergebnisse
1. Profilierung und Charakterisierung von circRNAs in Nierenbiopsien von LN-Patienten
Insgesamt wurden 18505 circRNA-Transkripte identifiziert, darunter 11411 hochregulierte und 7094 herunterregulierte circRNAs. Das hierarchische Cluster-Histogramm zeigte, dass die Expressionsniveaus der circRNAs zwischen LN-Nierenbiopsien und normalen Kontrollnieren gruppiert waren. 171 circRNAs wiesen signifikante Unterschiede in der Expression zwischen der LN-Gruppe und der normalen Kontrollgruppe auf.
 Abbildung 1. Die Profilierung und Eigenschaften von circRNAs. (A) Clustered Heatmap. Das Rot steht für hochregulierte circRNAs und das Grün für herunterregulierte circRNAs. (B) Vulkan-Diagramm. (C-F) Die Eigenschaften von unterschiedlich exprimierten circRNAs.
Abbildung 1. Die Profilierung und Eigenschaften von circRNAs. (A) Clustered Heatmap. Das Rot steht für hochregulierte circRNAs und das Grün für herunterregulierte circRNAs. (B) Vulkan-Diagramm. (C-F) Die Eigenschaften von unterschiedlich exprimierten circRNAs.
2. Das Interaktionsnetzwerk von CircRNAs und miRNAs
Das Interaktionsnetzwerk der CircRNAs/miRNAs wurde durch konservierte Seed-Matching-Sequenzen vorhergesagt. Es zeigte, dass alle 40 circRNAs ihre jeweiligen miRNA-Antwortelemente (MREs) enthielten. Die fünf wichtigsten miRNAs, die von jeder der 40 circRNAs reguliert werden, wurden als Netzwerk dargestellt (Abbildung 2).
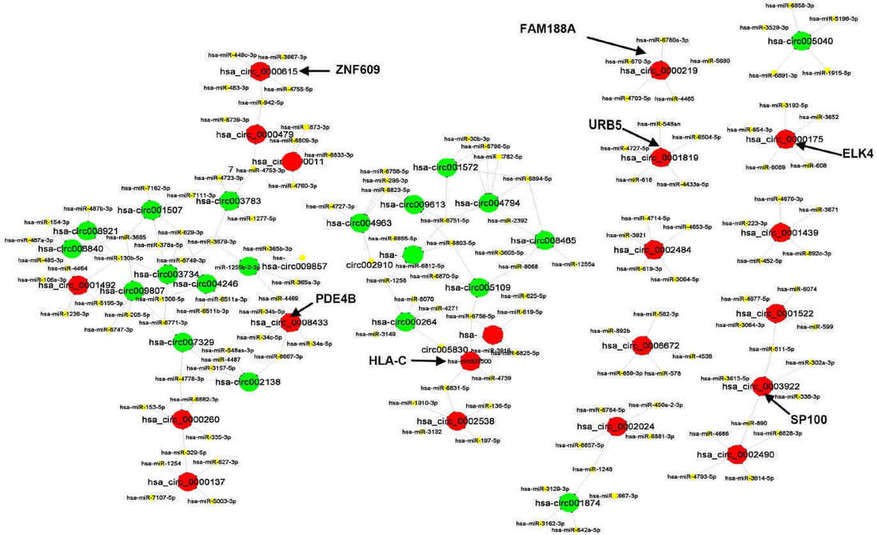 Abbildung 2. Das Interaktionsnetzwerk zwischen circRNAs und miRNAs.
Abbildung 2. Das Interaktionsnetzwerk zwischen circRNAs und miRNAs.
3. Vorhersage von Funktionen und Wegen
Die Funktionen von differentiell exprimierten circRNAs wurden durch die Analyse von GO und KEGG vorhergesagt. Die GO-Anreicherungsanalyse ergab, dass die funktionalen Rollen der Zielwirtgene mit biologischen Prozessen, zellulären Komponenten und molekularen Funktionen verknüpft waren. Die KEGG-Analyse identifizierte 23 Wege, die mit den Funktionen von 142 hochregulierten circRNAs in Zusammenhang standen, und sowohl der hypoxieinduzierbare Faktor-1 (HIF-1) Signalweg als auch der Neurotrophin-Signalweg waren an der Regulierung der Expression von NF-kB beteiligt.
 Abbildung 3. Vorhergesagte Funktionen der überexprimierten circRNAs in LP.
Abbildung 3. Vorhergesagte Funktionen der überexprimierten circRNAs in LP.
4. Wechselwirkung zwischen circHLA-C und miR-150
CircHLA-C korrelierte positiv mit den Parametern der Krankheitsaktivität bei LN. Und miR-150 zeigte eine Tendenz zur negativen Korrelation mit circHLA-C (Abbildung 4).
 Abbildung 4. Die Interaktion zwischen renalem circHLA-C und miR-150 in LN.
Abbildung 4. Die Interaktion zwischen renalem circHLA-C und miR-150 in LN.
Fazit
Die Autoren identifizierten 171 unterschiedlich exprimierte circRNAs und validierten sieben signifikant hochregulierte circRNAs in den Nieren von LN-Patienten der Klasse IV, wobei circHLA-C positiv mit der Krankheitsaktivität von LN korrelierte. circHLA-C war signifikant erhöht, während miR-150 verringert war, was eine negative Korrelation und eine perfekte Bindungssequenzübereinstimmung zeigte. Die Studie lieferte auch das erste Profiling von renalen circRNAs und ein Interaktionsnetzwerk zwischen circRNAs und miRs bei LN.
Referenz:
- Luan J, Jiao C, Kong W, u. a.circHLA-C spielt eine wichtige Rolle bei der Lupusnephritis, indem es miR-150 adsorbiert. Molekulare Therapie-Nukleinsäuren, 2018, 10: 245-253.
Verwandte Publikationen
Hier sind einige Veröffentlichungen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Das Merkelzell-Polyomavirus kodiert zirkuläre RNAs (circRNAs), die ein dynamisches circRNA/microRNA/mRNA-Regulationsnetzwerk ermöglichen.
Journal: Mbio
Jahr: 2020
Identifizierung von zirkulären RNAs, die die Proliferation von Kardiomyozyten in neugeborenen Schweineherzen regulieren
Journal: JCI Insight
Jahr: 2024
Zirkuläre DNA-Tumorviren erzeugen zirkuläre RNAs.
Zeitschrift: Mitteilungen der Nationalen Akademie der Wissenschaften
Jahr: 2018
KMT2A assoziiert mit dem PHF5A-PHF14-HMG20A-RAI1 Subkomplex in Stammzellen des Pankreaskarzinoms und reguliert epigenetisch deren Eigenschaften.
Zeitschrift: Nature Communications
Jahr: 2023
Krebsassoziierte DNA-Hypermethylierung von Polycomb-Zielen erfordert die duale Erkennung von Histon H2AK119-Ubiquitinierung und der sauren Tasche des Nucleosoms durch DNMT3A.
Journal: Wissenschaftliche Fortschritte
Jahr: 2024
Genomisches Imprinting-ähnliches monoalleles väterliches Ausdrucksmuster bestimmt das Geschlecht von Kanalkatzenfischen.
Journal: Wissenschaftliche Fortschritte
Jahr: 2022
Mehr ansehen Artikel, die von unseren Kunden veröffentlicht wurden.


