
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Mikrosatellitenentwicklung
CD Genomics bietet jetzt ein Next-Generation-Sequenzierung basierte Methode zur Entwicklung von Mikrosatellitenmarkern für Ihre Interessensart, um der wissenschaftlichen Gemeinschaft zugutekommen.
Die Einführung der Mikrosatellitenentwicklung
Mikrosatelliten, auch als SSR bezeichnet, bestehen aus einem wiederholten Motiv mit ein bis sechs Nukleotiden und sind in den meisten eukaryotischen Genomen weit verbreitet. Variationen in der Anzahl der Wiederholungen erzeugen unterschiedliche Allele. SSR weist eine hohe Polymorphie, genomische Spezifität, Häufigkeit und Kodominanz auf. Dies macht sie zu geeigneten molekularen Markern für molekular unterstützte Zucht, molekulare Phylogenetik und Populationsgenetik.
Next-Generation-Sequenzierung (NGS) wird kürzlich zur Entwicklung von Mikrosatellitenmarkern verwendet. Traditionelle Methoden, die auf Kapillarsequenzierung basieren, sind zeitaufwendig und komplex. Im Vergleich zu traditionellen Methoden, NGS hat die Vorteile von hoher Durchsatzrate, massiv gesteigerter Ausbeute und niedrigen Kosten. Es kann sogar nicht-modellorganismen ausreichend analysieren. Wir haben die Illumina-NGS-Technologie genutzt, um Millionen von kurzen Fragmentlesungen zu erzeugen, die mit bioinformatischen Werkzeugen gescreent wurden, um Primer zu identifizieren, die polymorphe Mikrosatellitenloci amplifizieren.
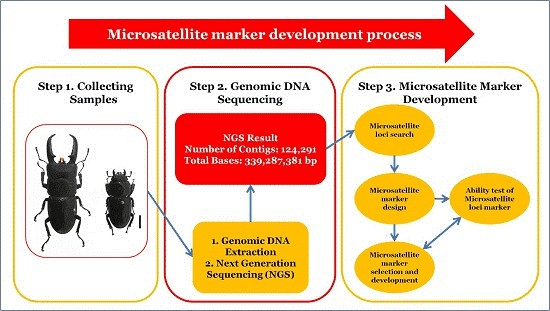
Der allgemeine Arbeitsablauf für die Entwicklung von Mikrosatelliten ist unten skizziert.
- DNA-Bibliotheksvorbereitung und Shotgun-Sequenzierung mit der Illumina-Plattform.
- Analyse der Sequenzierungsergebnisse mit bioinformatischen Werkzeugen und Identifizierung potenzieller Mikrosatellitenloci für die Primerentwicklung.
- Synthese von Primern und Auswahl von Mikrosatellitenloci durch Tests an mehreren Individuen zur Bewertung der Amplifikation und Polymorphismus.
- Analyse von Individuen, die wir mit nur den ausgewählten Primerpaaren genotypisieren möchten, die polymorphe Loci amplifiziert haben.
Wir werden mindestens 8 Proben an bis zu 60 Loci untersuchen, versuchen, die PCR-Optimierung für alle Loci durchzuführen, und schließlich werden wir unser Bestes tun, um 10 polymorphe Loci zu identifizieren, aber wir können nicht garantieren, da einige Populationen kein 'normales' Maß an Polymorphismus aufweisen.
Vorteile der Entwicklung von Mikrosatelliten
Die Anwendung von Hochdurchsatz-Sequenzierung (HTS) Die Entwicklung von Mikrosatelliten erhöht erheblich deren Effizienz und Präzision und bietet mehrere bemerkenswerte Vorteile gegenüber traditionellen Methoden:
- Massive Datenerfassung: HTS kann in kurzer Zeit ein beträchtliches Volumen an genomischen Sequenzdaten erzeugen, was den Zeitrahmen für die Entwicklung von Mikrosatelliten erheblich verkürzt.
- Automatisierung: Vom Sequenzieren bis zur Datenanalyse können zahlreiche Prozesse automatisiert werden, wodurch der Bedarf an manueller Intervention verringert wird.
- Umfassende Abdeckung: Mit dem Potenzial, den Großteil des Genoms abzudecken, einschließlich nicht-kodierender und niedrig komplexer Regionen, HTS erhöht die Umfänglichkeit der Mikrosatellitenidentifikation.
- Hohe Auflösung: HTS verbessert die Genauigkeit bei der Identifizierung der Wiederholungseinheiten und Mengen von Mikrosatelliten und verringert somit die Wahrscheinlichkeit von Fehlidentifikationen.
- Reduzierte Einzelinstanzkosten: Trotz der höheren anfänglichen Ausrüstungskosten haben fortschrittliche Entwicklungen in der Technologie die Sequenzierungskosten pro Instanz kontinuierlich gesenkt, was die großflächige Entwicklung von Mikrosatelliten wirtschaftlicher macht.
- Parallele Verarbeitung: HTS ermöglicht die gleichzeitige Verarbeitung mehrerer Proben, wodurch die Kosten pro Einheit Probe weiter gesenkt werden.
- Möglichkeit für mehrschichtige Analysen: HTS bietet nicht nur Mikrosatelliteninformationen, sondern kann auch für andere genomische Analysen (wie SNP-Erkennung, Genexpressionsanalyse usw.) eingesetzt werden und liefert Datenunterstützung für umfassende Forschung.
- Entdeckung neuer Mikrosatelliten: Im Vergleich zu traditionellen Methoden kann HTS mehr neue Mikrosatellitenmarker identifizieren und somit die Bibliothek der Mikrosatellitenmarker erweitern.
Anwendung der Mikrosatellitenentwicklung
Studie der genetischen Vielfalt und der Populationsstruktur: Durch die Nutzung von Hochdurchsatz-Sequenzierung Für die Identifizierung und Entwicklung zahlreicher Mikrosatellitenmarker ist es möglich, umfassende Analysen durchzuführen, die die genetische Struktur von Populationen, den Genfluss und die genetische Variation untersuchen. Dieser Prozess kann entscheidende Einblicke in die Populationsdynamik sowie in evolutionäre Prozesse bieten.
Kartierung genomischer Sequenzen und Lokalisierung quantitativer Merkmalsloci (QTL): Mikrosatellitenmarker spielen eine entscheidende Rolle beim Aufbau genetischer Verknüpfungskarten sowie bei der Lokalisierung quantitativer Merkmalsloci (QTL). Die Präzision und Dichte der Marker, die durch Hochdurchsatz-Sequenzierung verbessert wurden, haben die Anforderungen an die erreichbare Auflösung in der genomischen Kartierung erhöht.
Artenidentifikation und taxonomische Klassifikation: Mikrosatellitenmarker, die mit Hochdurchsatztechnologie entwickelt wurden, können effektiv in den Bemühungen zur Artenidentifikation und taxonomischen Forschung eingesetzt werden. Sie helfen bei der Erkennung genetischer Unterschiede zwischen verschiedenen Arten oder Populationen und unterstützen somit die Bemühungen um den Schutz und die Verwaltung der biologischen Vielfalt.
Forschung in der Evolutionsbiologie: Mikrosatellitenmarker, die durch Hochdurchsatz-Sequenzierungkann bei der Durchführung evolutionärer Studien über verschiedene Arten oder Populationen von Bedeutung sein. Sie können die evolutionäre Geschichte und die phylogenetischen Beziehungen der Studienobjekte offenlegen und somit unser Verständnis der Mechanismen hinter der biologischen Evolution bereichern.
Agrar- und Zuchtanwendungen: Im Bereich der Landwirtschaft und Zucht wurden Mikrosatellitenmarker entwickelt, die mithilfe von Hochdurchsatz-Sequenzierung kann zur Bestätigung der Rasseidentität, zur Auswahl in Zuchtprogrammen und zur Umsetzung genetischer Verbesserungen sowohl bei Pflanzen- als auch bei Tierarten eingesetzt werden. Dies erhöht die Effizienz und Genauigkeit landwirtschaftlicher und züchterischer Bemühungen.
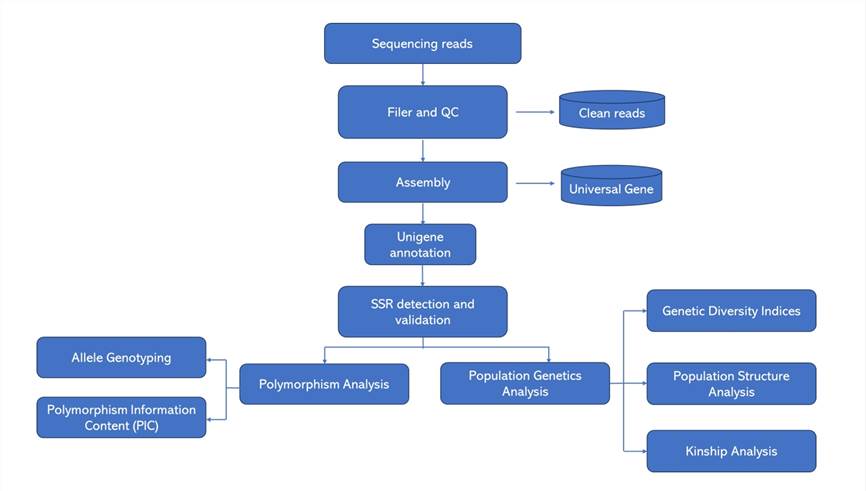
Mikrosatelliten-Entwicklungsworkflow
Die Umsetzung von Next-Generation Sequencing (NGS) Die Technologie im Anbau von Mikrosatelliten hat deren Effizienz, Präzision und Kosteneffektivität erheblich gesteigert. Durch einen systematischen Fortschritt, der die Probenvorbereitung umfasst, DNA-SequenzierungDurch Datenverarbeitung, Identifizierung von Mikrosatelliten, Primerdesign, Validierung des Primers und Testung von Polymorphismen können wir die Entwicklung von Mikrosatellitenmarkern schnell und effektiv fördern.
Dienstspezifikation
Musteranforderungen
|
|
 |
Sequenzierungsstrategien
|
 |
Datenanalyse
|
Analyse-Pipeline
Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
CD Genomics verpflichtet sich, Ihnen gebrauchsfertige, garantierte, getestete polymorphe SSRs anzubieten, und wir haben kein rechtliches Anrecht auf eines Ihrer SSR-Marker. Wenn Sie zusätzliche Anforderungen oder Fragen haben, zögern Sie bitte nicht, uns zu kontaktieren.
Referenz:
-
Feng S, He R, Lu J, u. a.Entwicklung von SSR-Markern und Bewertung der genetischen Vielfalt in medizinischen Chrysanthemum morifolium-Kultivaren. Grenzen der Genetik. 2016, 7:113.
Demo-Ergebnisse
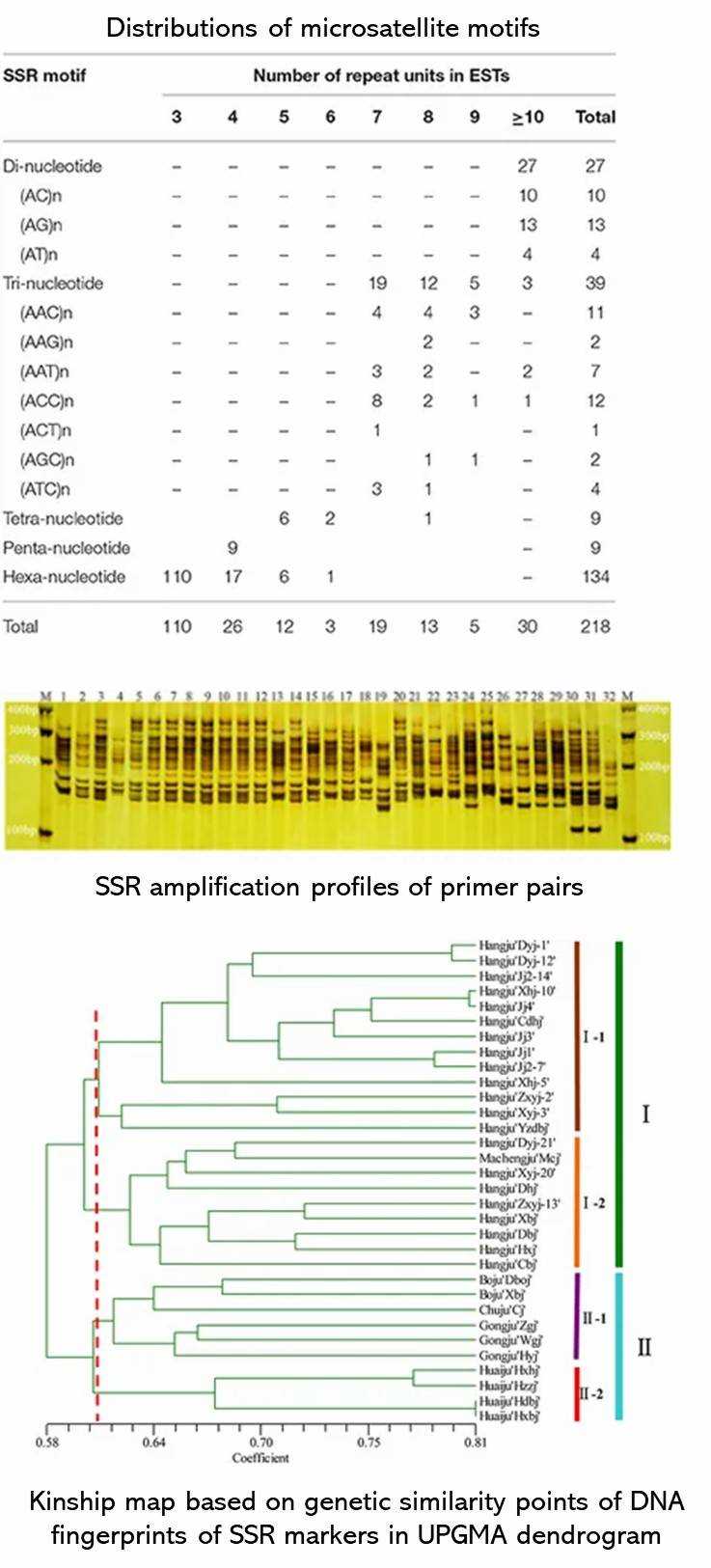 (Feng u. a.., 2016)
(Feng u. a.., 2016)
FAQs zur Entwicklung von Mikrosatelliten
1. Warum sind Mikrosatelliten in der genetischen Forschung wichtig?
Mikrosatelliten haben einen erheblichen Wert als genetische Marker in einer Vielzahl genetischer Untersuchungen, aufgrund ihrer hohen Variabilität, co-dominanten Vererbung und weit verbreiteten Präsenz in Genomen. Sie werden häufig in Studien über Populationsgenetikevolutionäre Biologie, forensische Untersuchungen, Vaterschaftstests und Verknüpfungsanalyse.
2. Wie werden Mikrosatelliten entwickelt?
Die Ableitung von Mikrosatelliten beginnt typischerweise mit der Hochdurchsatz-Sequenzierung von genomischer DNA, die einer bioinformatischen Analyse unterzogen wird, um sich wiederholende Sequenzen zu identifizieren. Anschließend werden die Sequenzen auf die Eignung für Mikrosatellitenloci überprüft, basierend auf entscheidenden Faktoren wie der Länge der Wiederholungsmotive, der Anzahl der Wiederholungen und den Eigenschaften der flankierenden Regionen.
3. Welche Faktoren sollten bei der Gestaltung von Mikrosatelliten-Primern berücksichtigt werden?
Die Gestaltung von Mikrosatellitenprimern erfordert die sorgfältige Berücksichtigung mehrerer wichtiger Parameter. Dazu gehören die Länge der Primer, die normalerweise zwischen 18 und 25 Nukleotiden liegt, die Schmelztemperatur (Tm), der Gehalt an Guanin-Cytosin-Paaren (GC-Gehalt) und die Spezifität gegenüber dem Ziel. Gemeinsam tragen diese Faktoren dazu bei, die erfolgreiche Amplifikation des Zielmikrosatellitenlokus durch die Polymerase-Kettenreaktion (PCR) sicherzustellen.
4. Wie werden Mikrosatellitenmarker validiert?
Die Validierung von Mikrosatellitenmarkern kann durch PCR-Amplifikation, die sich auf die gewünschten Loci konzentriert, erreicht werden, mit erzeugten Primern, die speziell für diese Aufgabe entworfen wurden. Dieser Schritt wird dann von Elektrophoreseanalysen gefolgt, die die erwarteten Größen der Amplifikate untersuchen und bestätigen. Eine anschließende Überprüfung auf Polymorphismus erfolgt, um die Unterschiede der Mikrosatellitenallele zwischen verschiedenen Organismen oder Populationen zu überprüfen.
5. Welche Herausforderungen sind mit der Entwicklung und Anwendung von Mikrosatelliten verbunden?
Der Entwicklungsprozess von Mikrosatelliten kann auf bestimmte Herausforderungen stoßen, wie zum Beispiel die Identifizierung geeigneter Loci, die einen hohen Grad an Polymorphismus aufweisen, sowie die Minderung des Risikos von Genotypisierung Fehler, die aus Stotterbändern oder Allelverlust resultieren können. Darüber hinaus können technische Herausforderungen im Zusammenhang mit der Übertragbarkeit zwischen Arten und der Entwicklung von Multiplex-PCR-Assays auftreten.
Mikrosatelliten-Entwicklungsfallstudien
Entwicklung neuer SSR-Marker für FlachsLeinsamen L.) Verwendung von reduzierter Repräsentationsgenomsequenzierung
Zeitschrift: Frontiers in Plant Science
Impactfaktor: 4,298
Veröffentlicht: 13. Januar 2017
Hintergrund
Die Flachszucht im Nordosten Chinas steht vor Herausforderungen bei der Entwicklung anpassungsfähiger Sorten mit verbessertem Ertrag und Qualität. Die markergestützte Selektion (MAS) bietet eine Lösung, wobei jüngste Fortschritte bei der Entdeckung von SSR-Markern vielversprechend sind. Die Nutzung von Next-Generation-Sequenzierung Methoden, zahlreiche SSR-Marker wurden schnell identifiziert, was Zuchtprogramme unterstützt. Die Illumina-Sequenzierungsplattform, die aufgrund ihrer hohen Durchsatzrate und Genauigkeit ausgewählt wurde, hat die systematische Identifizierung von SSR-Markern für den sofortigen Einsatz in der Flachszucht erleichtert.
Methoden
- 48 Sorten/Zugänge
- DNA-Vorbereitung
- Reduzierte Repräsentationsgenomsequenzierung (RRGs)
- Illumina-Sequenzierungsplattform
- Shotgun-Sequenzierung
- Identifizierung von SSRs
- SSR-Primerpaar-Design
- Genetischer Diversitätstest
Ergebnisse
Im Flachsgenom haben die Autoren 1.720 Loci für einfache Sequenzwiederholungen (SSR) identifiziert, von denen 1.574 neuartige Funde sind. Die SSR-Motivtypen bestehen überwiegend aus Trinukleotid- (56,1 %) und Dinukleotid- (35,23 %) Wiederholungen, basierend auf unseren Daten, die in Tabelle 1 aufgeführt sind.
Tabelle 1. Häufigkeiten verschiedener SSR-Wiederholungsmotivtypen.

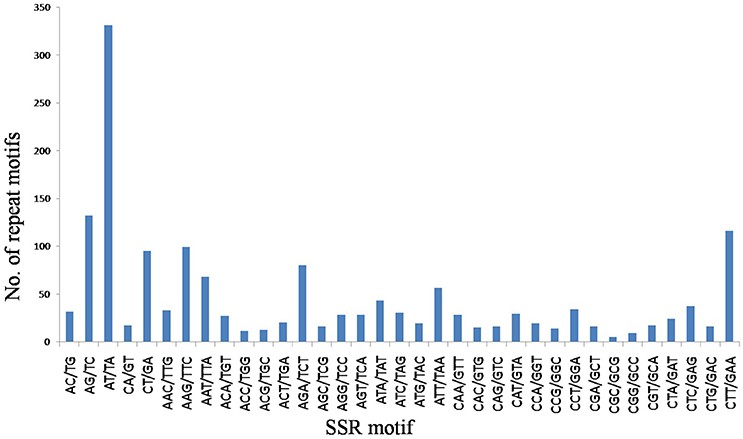 Abb. 1. Anzahl der Dinukleotid- und Trinukleotid-SSRs, klassifiziert nach ihren Motiven.
Abb. 1. Anzahl der Dinukleotid- und Trinukleotid-SSRs, klassifiziert nach ihren Motiven.
Mit 62 Primerpaaren wurde die Identifizierung von Polymorphismen an 48 Flachssorten aus Nordostchina durchgeführt, was ihre Klassifizierung hauptsächlich in Faser- und Ölsorten offenbarte (Abb. 2). Bemerkenswerterweise wiesen innerhalb der Fasersorten die Sorten "NEW1" und "Venus" deutlich unterschiedliche genetische Hintergründe auf. Ebenso unterschied sich der genetische Hintergrund von "A0529" von anderen Ölsorten. Folglich haben diese drei Flachssorten erhebliche Implikationen für die Flachszucht.
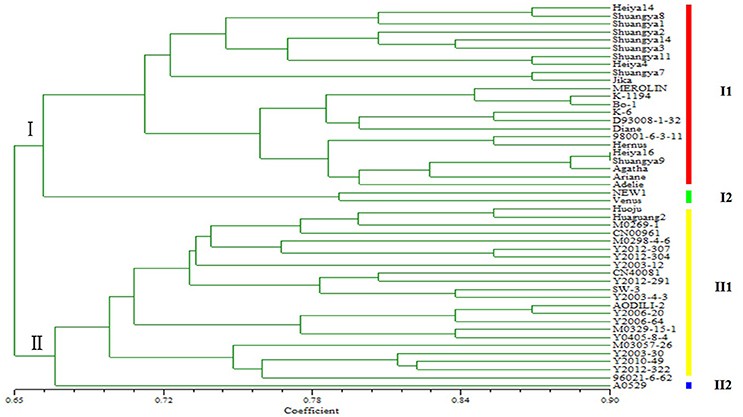 Abb. 2. Genetische Vielfalt von 48 Flachskultivaren/-zugängen basierend auf SSR-Markern.
Abb. 2. Genetische Vielfalt von 48 Flachskultivaren/-zugängen basierend auf SSR-Markern.
Fazit
Die Autoren entwickelten 1574 neuartige SSRs in Flachs mittels reduzierter Repräsentationsgenom-Sequenzierung. Davon wurden 62 SSR-Stellen für die Primer-Design ausgewählt, um die genetische Vielfalt in 48 Flachssorten zu bewerten. Die Ergebnisse zeigten eine deutliche Differenzierung zwischen Faser- und Leinsamenflachssorten. Diese SSRs werden entscheidend für die genetische Analyse und Kartierung in Flachs und anderen Kulturen sein.
Referenz:
- Wu J, Zhao Q, Wu G, u. a.Entwicklung neuartiger SSR-Marker für FlachsLeinsamen L.) unter Verwendung von reduzierter Repräsentationsgenom-Sequenzierung. Grenzen der Pflanzenwissenschaften. 2017(7):2018.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Pilze: Freunde oder Feinde – eine wissenschaftliche Outreach-Initiative zur Sammlung von luftgetragenen Pilzsporen durch Schüler der Oberstufe
Zeitschrift: Journal für Mikrobiologie und Biologieausbildung
Jahr: 2024
Kleine, aber signifikante genetische Differenzierung zwischen Populationen von Phyllachora maydis im Mittleren Westen der Vereinigten Staaten, aufgedeckt durch Mikrosatelliten (SSR)-Marker.
Journal: bioRxiv
Jahr: 2023
Das genetische Erbe von Fragmentierung und Übernutzung im bedrohten medizinischen afrikanischen Pfefferbaum, Warburgia salutaris
Journal: Wissenschaftliche Berichte
Jahr: 2020
Bewertung von Plasma-Biomarkern für die A/T/N-Klassifikation der Alzheimer-Krankheit bei Erwachsenen karibisch-hispanischer Ethnie
Journal: JAMA Netzwerk Offen
Jahr: 2023
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


