
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Genomsequenzierung
Was ist die Genom-DNA-Sequenzierung?
Die genomische DNA-Sequenzierung ist eine anspruchsvolle Methode, die auf überlegenen Hochdurchsatz-Sequenzierungsstrategien basiert, um das gesamte genomische DNA eines Organismus gründlich zu untersuchen. Das Hauptziel dieses wissenschaftlichen Vorgehens besteht darin, ein tiefes Verständnis der genetischen Informationen zu erlangen, die im Organismus eingeschlossen sind. Diese Technik hat eine außergewöhnliche Bedeutung in der biologischen Forschung und bietet eine solide datengestützte Grundlage für die Analyse des komplexen Zusammenhangs zwischen Genen, Krankheiten und Phänotypen. Der Einsatz der genomischen DNA-Sequenzierung verbessert unser Verständnis der biologischen Funktionalität eines Organismus, der zugrunde liegenden Mechanismen, die die Krankheitsentstehung antreiben, und des gerichteten Fortschritts der biologischen Evolution.
Was ist die Methode der genomischen DNA-Sequenzierung?
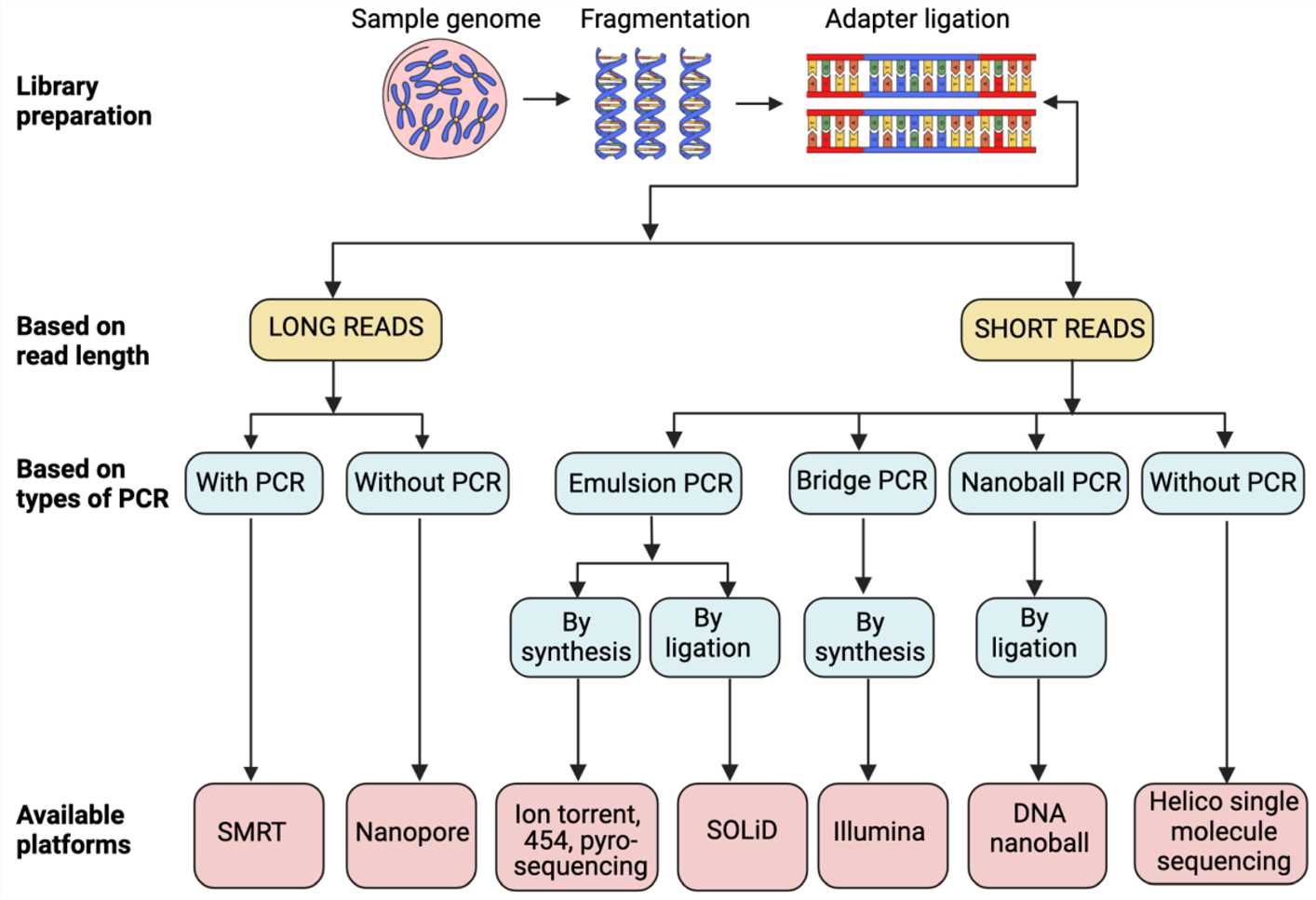
Derzeit können DNA-Sequenzierungstechnologien kurz in drei Hauptkategorien unterteilt werden. Die erste Kategorie umfasst die konventionellen Sanger-Sequenzierungstechnologie, allgemein als Sequenzierung der ersten Generation bezeichnet, wird weithin als der Goldstandard im klinischen Diagnostiksektor anerkannt. Die folgende Kategorie umfasst die Hochdurchsatzsequenzierung (HTS) oder Next-Generation-Sequenzierung (NGS) Technologien, die beide nachweislich in der Lage sind, ein enormes Volumen an DNA-Molekülen mit unvergleichlicher Effizienz schnell zu verarbeiten. Die letzte Kategorie umfasst Einzelmolekül-Sequenzierungstechnologien, die ohne die Abhängigkeit von PCR-Amplifikation direkt einzelne DNA-Moleküle sequenzieren können. Diese letzte Kategorie wird oft als Sequenzierungstechnologie der dritten Generation bezeichnet.
Sanger-Sequenzierung
Der Sanger-SequenzierungsmethodeEine archetypische Technik in der DNA-Sequenzierung basiert auf der engen Kopplung zwischen spezifischen Primern und Template-DNA-Molekülen. Während des Sequenzierungsprozesses katalysiert die DNA-Polymerase die progressive Addition der vier Typen von Desoxyribonukleotidtriphosphaten (dNTPs) an die an den Primer gebundene Template-DNA. Die Synthese neuer DNA-Stränge wird durch die Bildung kovalenter Bindungen zwischen dem 3'-Kohlenstoffatom eines Desoxyribosemoleküls und dem 5'-Kohlenstoffatom des nachfolgenden Nukleotids erleichtert. Dieser Prozess wird fortgesetzt, bis ein Terminator, ddNTP, erreicht wird, der am 3'-Ende ein Sauerstoffatom fehlt, was zur Beendigung der DNA-Strangsynthese führt.
NGS
Im Vergleich zur Sanger-Sequenzierungsmethode, Hochdurchsatz-Sequenzierungstechnologien wie die Illumina-Sequenzierung zeigen überlegene Effizienz, Durchsatz und Kosteneffektivität. Derzeit hat sie sich als gängige Methode in der modernen Genomforschung etabliert und findet breite Anwendung in verschiedenen Bereichen.
Nutzen ziehen aus NGS-Technologie hat erfolgreich die gleichzeitige Durchführung von Millionen von Sequenzierungsreaktionen ermöglicht, was einen bemerkenswerten technischen Durchbruch darstellt. In der Vergangenheit konnten zuverlässige Nukleotidsequenzen nur durch den koordinierten Betrieb von acht verschiedenen Reaktionsgemischen erhalten werden. Jetzt hingegen kann die Basensequenzinformation direkt während des synchronen Prozesses der Sequenzverlängerung und -erkennung identifiziert werden.
Mit dem Aufkommen der NGS-Technologie hat sich der Anwendungsbereich der Genomik erheblich erweitert. Derzeit ist die DNA-Sequenzierung ein integraler Bestandteil in mehreren Bereichen, einschließlich Grundlagenforschung, translationale Forschung, medizinische Diagnostik und forensische Wissenschaft. Trotz der bemerkenswerten Erfolge der NGS-Technologie bei der Kosten- und Zeitreduktion trägt die relativ kurze "Lese-Länge", die sie erzeugt, zu den hohen rechnerischen Anforderungen für die anschließende Genomassemblierung bei. Dennoch sind wir optimistisch, dass diese Herausforderungen mit kontinuierlichem technologischen Fortschritt und Optimierung allmählich gelöst werden.
 Überblick über verschiedene NGS-Technologien (Heena Satam et al., Biologie 2023)
Überblick über verschiedene NGS-Technologien (Heena Satam et al., Biologie 2023)
Einzelmolekül-Sequenzierung
Einzelmolekül-Sequenzierung, auch bekannt als Langzeit-Sequenzierung, gewinnt zunehmend die Aufmerksamkeit der wissenschaftlichen Gemeinschaft aufgrund ihrer Überlegenheit im Bereich des Lesens langer Sequenzen. Technologien dieser Art fallen hauptsächlich in zwei Kategorien: Einzelmolekül-Echtzeit (SMRT) Sequenzierung und Nanoporen-Sequenzierung.
Unsere DNA-Sequenzierungsdienste
Ausgestattet mit fortschrittlichen NGS-Plattformen, modernster Technologie und unterstützt von spezialisierten Wissenschaftlern bietet CD Genomics eine breite Palette genomischer Lösungen, um Ihre vielfältigen Forschungsziele und Budgets zu erfüllen.
-
Whole Genome Sequenzierung
-
Die gesamte Genomsequenzierung (WGS), eine revolutionäre Technik, wird nun weitreichend in der Forschung zu menschlichen und tierischen Genomen eingesetzt. Ihre grundlegende Aufgabe besteht darin, die vollständige Genomsequenz innerhalb einer biologischen Zelle umfassend zu untersuchen und zu ordnen, wobei alle Arten von Mutationen vom ersten bis zum letzten DNA-Abschnitt sorgfältig erfasst werden. Dies hat erhebliche Auswirkungen auf unser Verständnis der genetischen Informationen eines Organismus, der Krankheitsmechanismen und der Beziehungen zwischen Genen und Merkmalen.
Die Entwicklung und Anwendung der gesamten Genomsequenzierung gehen über den Menschen hinaus in andere biologische Bereiche. Bei Organismen, die über keine geeigneten Referenzgenome oder über Referenzgenome von geringer Qualität verfügen, von neuem Sequenzierung und Assemblierungstechniken sind äußerst wertvoll. Durch WGS können Forscher die vollständigen Genominformationen eines Organismus erhalten, was wichtige Grundlagen für weitere Untersuchungen zu Genfunktionen, Genom-Evolution, Genregulationsnetzwerken und mehr bietet.
In praktischen Anwendungen hat die Ganzgenomsequenzierung weltweit anerkannte Erfolge erzielt. Zum Beispiel hat WGS erfolgreich die Genomsequenzen verschiedener Tiere und Pflanzen entschlüsselt und bietet damit umfassende Unterstützung für die Forschung in Bereichen wie Landwirtschaft und Medizin. Darüber hinaus spielt WGS eine entscheidende Rolle bei der Erkennung pathogener Mikroben, der forensischen Genetik und Studien zur Biodiversität, unter anderem.
-
-
Whole Exome Sequencing: Gesamtes Exom-Sequenzierung
-
Innerhalb des menschlichen Genoms beträgt die Anzahl der Exons ungefähr 180.000, was 1-2% des gesamten genomischen Entitäts ausmacht, etwa 30 MB in rechnerischen Begriffen. Pathogene Mutationen in den protein-kodierenden Regionen des menschlichen Genoms machen etwa 85% der gesamten pathologischen Veränderungen aus. Die Ganzexomsequenzierung (WES) ist eine wichtige Technik, die die Amplifikation von DNA-Sequenzen aus Exonregionen durch Sondenhybridisierung begünstigt, bevor hochdurchsatz Sequenzierungsmethoden angewendet werden. Das Hauptziel hierbei ist es, genetische Mutationen zu identifizieren und zu untersuchen, die mit Krankheiten und evolutionären Metriken in kodierenden und regulatorischen Regionen (Untranslatierte Regionen, UTR) assoziiert sind. Die Zusammenstellung dieser Daten mit öffentlich verfügbaren Exomdaten unterstützt die tiefere Interpretation der Beziehung zwischen verschiedenen Mutationen und den daraus resultierenden Krankheitsmechanismen.
Im Vergleich zu WGS bietet die Whole Exome Sequencing mehrere Vorteile: a) Kosten-Effektivität: Im Vergleich zu WGS bietet die Whole Exome Sequencing eine überlegene Abdeckungstiefe und verbesserte Datenpräzision, was sie zu einer wirtschaftlich bevorzugten Wahl macht; b) Sequenzierungstiefe: Die Sequenzierungstiefe kann über 120x erreichen; c) Hohe Durchsatzkapazität: WES eignet sich hervorragend für großangelegte Studien mit zahlreichen Zielregionen; d) Hohe Präzision: Eine umfassende Sequenzierungsabdeckung geht mit hoher Datenakkuratheit einher, was gut optimierte Ergebnisse liefert.
-
-
Gezielte DNA-Sequenzierung / (Panel-Sequenzierung)
-
Targeted Resequenzierung ist eine Technik, die hauptsächlich Multiplex-Amplikon-Sequenzierung und Hybrid-Capture-Sequenzierung umfasst. Sie isoliert spezifische Gene oder genomische Regionen zur Sequenzierung. Im Vergleich zu WGS und WES weist die gezielte Resequenzierung folgende Vorteile auf:
Es ermöglicht eine hochpräzise Sequenzierung von wichtigen Genen mit einer tiefen Sequenzierung von über 500x, was zu einer genauen Identifizierung seltener Variationen führt.
Es ist wirtschaftlich effizient und erleichtert das Studium von krankheitsassoziierten Genen.
In der Lage, Variationen der Allelfrequenzen von bis zu 5% zu identifizieren.
Während einer einzelnen Detektion kann eine vertrauenswürdige Identifizierung von erblichen Mutationen erreicht werden.
-
-
Mitochondriale DNA (mtDNA) Sequenzierung
-
Mitochondriale DNA (mtDNA) ist ein Molekül im Zytoplasma von Zellen, dessen Struktur, Standort und Menge einen direkten Einfluss auf die physiologischen Funktionen und das Schicksal der lebenden Zelle ausüben. Die Sequenzierungstechnologie des mitochondrialen DNA ist eine revolutionäre Biotechnologie, die gezielt die Sequenzierung von mtDNA analysiert - dem entscheidenden Organell, das für den Energiestoffwechsel innerhalb der Zelle verantwortlich ist.
Durch die Offenlegung einer komplexen Karte der DNA-Zusammensetzung innerhalb einer Zelle ermöglicht die Sequenzierung von mitochondrialer DNA eine präzise Quantifizierung und Analyse von DNA-strukturellen Eigenschaften. Diese Technologie zeichnet ein detailliertes Porträt der phänotypischen Merkmale von Zellen und eröffnet damit einen neuen Forschungsweg für Wissenschaftler. Die Technologie bietet Forschern ein genaues und schnelles Mittel, um tief in die innere DNA-Struktur und Funktionalitäten der Zelle einzutauchen. Noch wichtiger ist, dass sie eine Schlüsselrolle bei der Aufdeckung der Geheimnisse spielt, die mit der genetischen Struktur von Arten und deren Reaktionen auf Umweltveränderungen verbunden sind.
Daher bietet die Sequenzierungstechnologie der mitochondrialen DNA den Forschern nicht nur eine effizientere Forschungsmethodik, sondern dient auch als wertvoller Bezugspunkt im Bereich der menschlichen Gesundheit und der medizinischen Forschung.
-
-
Menschliche mitochondriale DNA-Sequenzierung
-
Mitochondrien, integrale Organellen, die in eukaryotischen Zellen vorkommen, sind verantwortlich für die Kodierung von Genen, die mit ihrer Funktion in Verbindung stehen, und nehmen aktiv an zahlreichen Lebensprozessen teil. Die menschliche mitochondriale DNA, die durch ihre einzigartigen Eigenschaften und die kompakte, doppelsträngige, zirkuläre Struktur gekennzeichnet ist, hat die wissenschaftliche Gemeinschaft fasziniert.
Die menschliche mitochondriale DNA, die etwa 16 Kilobasenpaare lang ist, kodiert eine Vielzahl von Genen, um ihre grundlegende Rolle innerhalb der Zelle zu unterstützen. Ihre strukturelle Einfachheit in Verbindung mit den hochkonservierten kodierenden Regionen deutet auf eine evolutionäre Stabilität ihrer genetischen Sequenz hin, was die funktionale Forschung erheblich erleichtert.
Die maternale Vererbung der mitochondrialen DNA verleiht ihr einen einzigartigen Status in der genetischen Forschung. Da mitochondriale DNA überwiegend von der Mutter an den Nachwuchs weitergegeben wird, hat sie einen entscheidenden Wert in der Untersuchung von Erbkrankheiten. Darüber hinaus unterstreicht die schnelle Evolutionsrate der mitochondrialen DNA ihre zentrale Rolle im Prozess der biologischen Evolution.
Gleichzeitig deutet die niedrige Rekombinationsrate der mitochondrialen DNA darauf hin, dass sie einen hohen Wert in der Forschung zu Genmutationen und genetischer Variation hat. Bemerkenswert ist, dass die hohe Kopienzahl der mitochondrialen DNA ihre wesentliche Funktion innerhalb der Zelle belegt.
Um tiefer in die Feinheiten der mitochondrialen DNA einzutauchen, setzen Forscher die Technologie der Flüssigkeits-Hybridisierungs-Probenfänge ein, um mitochondriale DNA anzureichern und die Hochdurchsatz-Sequenzierungsforschung zu erleichtern. Diese leistungsstarke Methodik entschlüsselt Aspekte der mitochondrialen DNA und legt ein solides Fundament für das Verständnis der unentbehrlichen Rolle der Mitochondrien im Theater der Lebensaktivitäten.
-
-
Chloroplast-DNA (cpDNA) Sequenzierung
-
Chloroplasten gehören zu den wichtigsten und verbreitetsten Organellen in Pflanzenzellen und dienen als zentraler Ort für die Photosynthese. Die Struktur- und Sequenzinformationen von Chloroplastengenomen sind von erheblichem Wert, um die Ursprünge, evolutionären Veränderungen und phylogenetischen Beziehungen verschiedener Arten aufzudecken. Gleichzeitig zeigt die Chloroplastentransformationstechnologie ein beträchtliches Potenzial für genetische Verbesserungen und die Produktion von bioaktiven Verbindungen, wobei die Struktur- und Sequenzanalyse von Chloroplastengenomen das Fundament dieses Transformationsprozesses bildet.
Traditionell bestand der Erwerb des Chloroplastengenoms einer Pflanze darin, degenerierte Primer unter Verwendung der konservativen Sequenzen des Chloroplastengenoms zu entwerfen, unbekannte Sequenzen zu amplifizieren und eine lange PCR-Amplifikation durchzuführen. Dieses amplifizierte Produkt wird einer Sanger-Sequenzierung unterzogen, und die Sequenzen werden dann zusammengefügt, um das vollständige Chloroplastengenom zu erhalten. Dieser Prozess ist jedoch oft zeitaufwendig.
Mit der Entwicklung wissenschaftlicher Technologien und dem Aufkommen neuer Sequenzierungswerkzeuge haben zahlreiche Forscher kürzlich Begeisterung für Hochdurchsatz-Sequenzierung gezeigt. Bei diesem Ansatz werden zunächst Chloroplasten isoliert, gefolgt von der Extraktion von cpDNA. Basierend auf ausgewählten Referenzen des Chloroplastengenoms wird dann Software verwendet, um diese Sequenzen zusammenzufügen, was letztendlich ein vollständiges Chloroplastengenom ergibt. Dennoch ist dieser Ansatz möglicherweise nicht auf alle Arten anwendbar. Zum Beispiel enthalten die Blätter höherer Pflanzen oft hohe Mengen an Pigmenten und Tanninen, was die Isolation von Chloroplasten und die Extraktion von cpDNA erschwert.
Je nach den spezifischen Anforderungen der Zielart kann eine totale DNA-Extraktion oder eine cpDNA-Extraktion durchgeführt werden. Informationen zum taxonomischen Status werden verwendet, um nach Referenz-Mitochondriensequenzen zu suchen, und degenerierte Primer werden entworfen, um PCR-Baiting, Amplifikation unbekannter Sequenzen und lange PCR-Amplifikation durchzuführen. Letztendlich wird die vollständige Chloroplastengenomsequenz durch eine Kombination aus Hochdurchsatz-Sequenzierung und Sanger-Sequenzierung erlangt.
-
-
Vollständige Plasmid-/Phagen-Sequenzierung
-
Plasmide und Phagen sind wichtige Werkzeuge in der modernen genetischen Forschung, die häufig für Klonierung, Vektorkonstruktion und synthetische Biologie verwendet werden. Unser Vollständige Plasmid-/Phagen-Sequenzierung Der Service ermöglicht eine vollständige, hochauflösende Analyse dieser genetischen Elemente, die Forschern hilft, Konstrukte zu validieren und Mutationen mit Vertrauen zu erkennen.
Im Gegensatz zur partiellen Sequenzierung oder Restriktionsmapping bietet die vollständige Sequenzierung von Plasmiden/Phagen eine vollständige Abdeckung – einschließlich Insert, Backbone und regulatorischen Regionen – was sie ideal für Anwendungen wie Gentherapie, Impfstoffentwicklung und mikrobielle Ingenieurwissenschaften macht. Die Fähigkeit, die Integrität der Sequenz zu bestätigen, ist besonders entscheidend für regulatorische Einreichungen und nachgelagerte funktionale Studien.
Während große oder stark repetitive Plasmide herausfordernd sein können, kombiniert unsere Plattform Langzeit-Sequenzierung mit fortschrittlichen Fehlerkorrektur-Pipelines, um eine genaue Assemblierung und Annotation zu gewährleisten – selbst bei komplexen Konstrukten.
Warum es wichtig ist:
Validiert die vollständige Plasmid-/Phagenstruktur, nicht nur die Einsätze.
Erkennt Mutationen, Umstellungen und Off-Target-Modifikationen.
Unterstützt Anwendungen in der synthetischen Biologie, Gentherapie und Impfstoffdesign.
-
-
TCR/BCR-Sequenzierung (TCR/BCR-Seq)
-
T-Zell-Rezeptoren (TCRs) und B-Zell-Rezeptoren (BCRs) bestimmen, wie das Immunsystem Bedrohungen erkennt und darauf reagiert. Unser TCR/BCR-Sequenzierung Service-Profile das gesamte Repertoire dieser Immunrezeptoren und zeigt Muster der Vielfalt, klonale Expansion und antigen-spezifische Reaktionen auf.
Durch die Erfassung der variablen Regionen von TCR- und BCR-Transkripten ermöglicht diese Methode Forschern, die Immun-Dynamik bei Krankheiten, Immuntherapien und der Impfstoffentwicklung zu verfolgen. Von tumorinfiltrierenden Lymphozyten bis hin zu autoimmunen Signaturen liefern die Daten entscheidende Einblicke in den Immunstatus und die Funktion.
Was unseren Service auszeichnet, ist die Kombination aus tiefem Sequencing, dualer Kettenrekonstruktion (z. B. TCRα/β oder IgH/IgL) und maßgeschneiderter Bioinformatik, die hypervariable Regionen mit hoher Sensitivität verarbeitet.
Anwendungen umfassen:
Überwachung von T-Zell- und B-Zell-klonalen Expansionen bei Krebs und Infektionen
Charakterisierung von Immunrepertoires als Reaktion auf Immuntherapie
Untersuchung der Immun-Dysregulation bei Autoimmunität und Transplantation
-
-
Amplicon-Sequenzierung
-
Die Amplicon-Sequenzierung ist eine leistungsstarke Technik, die spezifische universelle Primer verwendet, um variable Regionen der 16S rDNA/18S rDNA/ITS oder funktionalen Gene von Mikroben in unterschiedlichen Umgebungen zu amplifizieren. Anschließend untersuchen wir durch Hochdurchsatz-Sequenzierung die Sequenzvariationen und Abundanzinformationen der Produkte der Polymerase-Kettenreaktion (PCR). Dieser Ansatz erleichtert die Analyse von Diversitäts- und Verteilungsmustern mikrobieller Gemeinschaften in bestimmten Umgebungen und enthüllt somit die relative Abundanz und evolutionären Beziehungen zwischen den zahlreichen Mikrobenarten, die in Umweltproben vorkommen.
-
-
Virusgenomsequenzierung
-
Die vollständige Virusgenomsequenzierung (VWGS) umfasst eine umfassende Analyse der genomischen Sequenz von Viren mittels zweiter und dritter Generation von Sequenzierungsplattformen. Durch die Nutzung bioinformatischer Methoden interpretiert dieser Ansatz kodierende Informationen und führt eingehende Untersuchungen zu viralen pathogenen Systemen und der evolutionären Entwicklung ihrer Genome durch. Disziplinen aus der strukturellen Genomik und der vergleichenden Genomik, einschließlich differentieller Analyse, homologer Genanalyse, Kollinearitätsanalyse und Analyse der Speelev evolution, wenden solche Techniken an, um diese Aspekte genau zu untersuchen. Solche Bemühungen verbessern unser Verständnis von viraler Vielfalt, Ökologie, Anpassungsfähigkeit und evolutionären Mustern und helfen, das Auftreten von aufkommenden Infektionskrankheiten vorherzusagen.
Forschung zum Chloroplastengenom hat eine erhebliche Bedeutung in den Lebenswissenschaften. Seine Einzigartigkeit zeigt sich in der Aufdeckung entscheidender Fragen wie der Herkunft und Evolution von Arten, und seine Anwendbarkeit erstreckt sich auf andere Disziplinen wie die Landwirtschaft. Mit der rasanten Entwicklung von Hochdurchsatz-Sequenzierungstechnologien ist die Untersuchung von Chloroplasten zu einem leistungsstarken Werkzeug geworden, um die Herkunft, Struktur und evolutionären Fragen zellulärer Organellen zu erforschen. Durch die Nutzung von Sequenzierungsplattformen der zweiten und dritten Generation, die Durchführung von Hochdurchsatz-Sequenzierungen an pflanzlichen Chloroplasten sowie die Durchführung von Tiefensequenzierungen und bioinformatischen Analysen können wertvolle Informationen über die Chloroplastengenomsequenz, kodierende Gene und genetische Evolution gewonnen werden.
-
-
Lange Amplicon-Analyse (LAA)
-
Im Bereich der genetischen Forschung hat die Long Amplicon Analyse (LAA) erhebliche Aufmerksamkeit als eine äußerst effiziente und praktische Methode zur Genuntersuchung erlangt. LAA, eine Technik, die auf der PCR-Technologie basiert, richtet sich hauptsächlich auf die Amplifikation gezielter Gensequenzen durch das Design spezifischer Primer. Im Gegensatz zur traditionellen PCR-Amplifikation entwirft LAA Primer für die distalen Bereiche der Zielsequenzen, was die effiziente Amplifikation langer DNA-Fragmente ermöglicht. Diese Methode zeichnet sich durch hohe Amplifikationseffizienz und Genauigkeit aus und reduziert effektiv falsche Amplifikationen aufgrund übermäßiger PCR-Zyklen.
LAA kann eingesetzt werden, um hochgradig abgedeckte genomische DNA-Fragmente zu erhalten, die entscheidend sind, um die Beziehung zwischen genomischer Struktur und Funktion zu entschlüsseln. Durch die Amplifikation spezifischer Genfragmente kann LAA effizient die Genexpressionsniveaus in verschiedenen Proben nachweisen und bietet eine Grundlage für die Diagnose und Behandlung von Krankheiten. Darüber hinaus kann LAA zur Erkennung von Mutationsstellen verwendet werden, was technische Unterstützung für die molekulare Diagnose genetischer Erkrankungen und Tumoren sowie anderer Bedingungen bietet. Zusätzlich kann LAA bei der Analyse von regulatorischen Elementen wie Promotoren und Enhancern dienen und somit die zugrunde liegenden Mechanismen der Regulierung der Genexpression untersuchen.
-
-
Oberflächliche Ganzgenomsequenzierung
-
Shallow Whole Genome Sequencing (sWGS) gehört zu den robusten und kosteneffizienten genomischen Testmethoden, die entwickelt wurden, um Variationen und die damit verbundenen genetischen Informationen im Genom eines Individuums zu erläutern. Per Definition bedeutet sWGS eine partielle Abdeckungssequenzierung der genomischen DNA, mit einer eher verdünnten Tiefe und Abdeckungsreichweite im Vergleich zur tiefen Whole Genome Sequencing. In praktischen Anwendungen ermöglicht das zufällige Fragmentieren der genomischen DNA, gefolgt von der Sequenzierung dieser Segmente, einen selektiven Zugang zu genomischen Informationen. Die Analyse dieser Informationsuntergruppen macht es möglich, neuartige Mutationen – Kopienzahlvariationen, epigenetische Variationen und mehr – zu erkennen.
Die Einführung von Hochdurchsatz-Sequenzierungsplattformen wie Illumina, PacBio und Oxford Nanopore hat eine schnelle Entwicklung der sWGS ermöglicht. Mit einem hohen Sequenzierungsdurchsatz und einer hohen Genauigkeit sind diese Plattformen in der Lage, massive Mengen an Proben in verkürzten Zeitrahmen zu sequenzieren. Darüber hinaus hat die fortschreitende Verfeinerung von Analysewerkzeugen für Sequenzierungsdaten, einschließlich Software-Suiten wie Bismark und CMap, den Prozess der Verwaltung und Analyse von Sequenzierungsdaten erheblich optimiert und den Forschern präzise Ergebnisse zur Mutationsdetektion geliefert.
In der biomedizinischen Forschung ist sWGS von unschätzbarem Wert. Durch den Vergleich der Genome von Krankheitsbetroffenen und gesunden Kontrollen können bedeutende Erkenntnisse über krankheitsassoziierte Genmutationen gewonnen werden, die eine solide Grundlage für die genetische Diagnostik und Gentherapie bieten. Diese Technik ermöglicht zudem genotypische und phänotypische Analysen von Organismen, wodurch die Dynamik zwischen Genen und ihrer Umwelt offenbart wird.
-
-
Zirkulierende Tumor-DNA (ctDNA) Sequenzierung
-
Zirkulierende Tumor-DNA (ctDNA) bezieht sich auf DNA-Fragmente, die von Tumorzellen in den Blutkreislauf freigesetzt werden. Die Sequenzierung dieser Fragmente ermöglicht es uns, genotypische Mutationsinformationen des Tumors zu ermitteln, was eine wesentliche Grundlage für die Tumorforschung bietet.
Der Hauptvorteil der ctDNA-Sequenzierung liegt in ihrer hohen Sensitivität und Spezifität. Im Vergleich zu herkömmlichen Gewebe-Biopsien kann die ctDNA-Sequenzierung Tumormutationen in einem früheren Stadium nachweisen, selbst wenn die Tumorgröße noch relativ klein ist. Das Spektrum der Anwendungen der ctDNA-Sequenzierung ist breit und umfasst die Identifizierung von Tumortypen, molekulare Subtypisierung, Auswahl gezielter Therapeutika, Wirksamkeitsüberwachung und Prognosebewertung, unter anderem.
Dennoch steht die ctDNA-Sequenzierung vor mehreren Herausforderungen. Erstens ist die Konzentration von ctDNA im Körper relativ niedrig, was hochsensible Nachweismethoden und -technologien erforderlich macht. Zweitens erhöht die Mischung von DNA aus normalen und Tumorzellen innerhalb der ctDNA-Sequenzen die Komplexität bei der Mutationsdetektion und Datenanalyse. Darüber hinaus erfordert die Interpretation der Ergebnisse der ctDNA-Sequenzierung fortlaufende Forschung und angesammelte Erfahrungswerte.
-
Vorteile unserer DNA-Sequenzierungsdienste
Einhaltung der höchsten Standards bei der Bereitstellung umfassender Genomiklösungen.
Expertise in verschiedenen Aspekten der Sequenzierung wie Versuchsdesign, Konstruktion von Zielanreicherungsbibliotheken und maßgeschneiderte bioinformatische Analysen.
Lieferung von schnellen, genauen, zuverlässigen und erschwinglichen Sequenzierungsdiensten.
Fähigkeit, Genomsequenzierungsdienste für eine Vielzahl von Proben anzubieten, einschließlich Mensch, Maus, Pflanze, Tier und Mikroben.
Mission, die Genomforschung zu fördern, indem der Zugang zu den neuesten Technologien auf diesem Gebiet bereitgestellt wird.
Flexibilität, Dienstleistungen an die individuellen Projektbedürfnisse anzupassen und speziell zugeschnittene Analysen bereitzustellen.
Ein beratender Ansatz zur Identifizierung der besten und wirtschaftlichsten Lösungen zur Erfüllung spezifischer Forschungsbedürfnisse.
Identification of factors required for m6A mRNA methylation in Arabidopsis reveals a role for the conserved E3 ubiquitin ligase HAKAI
New phytologist | 2017Distinct functions of wild-type and R273H mutant Δ133p53α differentially regulate glioblastoma aggressiveness and therapy-induced senescence
Cell Death & Disease | 2024High-Density Mapping and Candidate Gene Analysis of Pl18 and Pl20 in Sunflower by Whole-Genome Resequencing
International Journal of Molecular Sciences | 2020Generation of a highly attenuated strain of Pseudomonas aeruginosa for commercial production of alginate
Microbial Biotechnology | 2019

