
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
2b-RAD
CD Genomics bietet jetzt 2b-RAD-Sequenzierung an, eine neuartige Methode zur reduzierten Repräsentation der gesamten Genomsequenzierung und zur restriktionsstellenassoziierten DNA (RAD)-Sequenzierung für die Verknüpfungskartierung und Bestimmung von genomweiten Varianten auf kosteneffiziente Weise.
Was ist die 2b-RAD-Methode?
Die 2b-RAD (IIB-RAD) Methode, die erstmals 2012 von Wang et al. beschrieben wurde, stellt einen modernen Ansatz in der Hochdurchsatz-Genomik dar. SNP-Genotypisierung. Durch die Verwendung von Typ IIB Restriktionsendonukleasen, wie Bcg I, erzeugt diese Technik einheitliche DNA-Fragmente mit einer Länge von 33 bis 36 Basenpaaren, indem sie an spezifischen Erkennungsstellen schneidet. Dies beseitigt die Notwendigkeit für nachfolgende Größenauswahl-Schritte. Einer der Hauptvorteile der 2b-RAD-Methodik ist ihre Fähigkeit, konsistente Fragmentlängen über eine Vielzahl von Proben hinweg zu erzeugen, wodurch präzises und effizientes Sequenzieren sowie die anschließende SNP-Entdeckung erleichtert werden. Obwohl die 2b-RAD-Methode im Vergleich zu Einzel-Enzym-RAD-Ansätzen weniger Fragmente erzeugt, kompensiert sie dies mit überlegener Auflösung und Abdeckung. Folglich adressiert diese Technik mehrere inhärente Einschränkungen früherer Methoden wie RAD-Seq und GBSund bietet eine robuste Alternative für umfassende SNP-Analysen.
Einführung in 2b-RAD
Die 2b-RAD-Methode verwendet Typ IIB Restriktionsenzyme (REs), wie Bcg I, die genomische DNA auf beiden Seiten ihrer Erkennungsstellen spalten, um DNA-Fragmente doppelsträngiger DNA (dsDNA) mit festen Größen und überstehenden, nicht-kohäsiven Enden zu erzeugen. Anschließend werden diese DNA-Fragmente mit einem biotinylierten Adapter, der spezifisch für das ursprüngliche Enzym ist, erfasst. Im Vergleich zu ähnlichen Methoden wie der restriktionsstellenassoziierten DNA (RAD) und der Genotypisierung durch Sequenzierung (GBS) bietet die 2b-RAD-Technik eine überlegene Spezifität für die interessierenden Regionen, während die Erzeugung von Sequenzen aus nicht-informativen und repetitiven Regionen minimiert wird.
Die konsistente Generierung von kurzen und einheitlichen Tags durch die 2b-RAD-Methode bietet mehrere Vorteile, darunter die gezielte Ansprache aller Restriktionsstellen, eine gleichmäßige Sequencing-Tiefe über die Stellen hinweg, hohe Reproduzierbarkeit für quantitative Messungen, reduzierte Sequencing-Kosten pro Tag und eine verringerte Empfindlichkeit gegenüber DNA-Abbau. Darüber hinaus, um die verbesserten Sequencing-Kapazitäten zu nutzen von Next-Generation Sequencing (NGS) Auf den Plattformen haben wir ein fortschrittliches Protokoll integriert, das die Vorbereitung von fünf verketteten isoRAD-Tags für die Illumina-Paarendsequenzierung (PE) mit 150 bp ermöglicht. Dieses Protokoll bietet Forschern größere Vielseitigkeit und Effizienz bei der Gestaltung von Bibliothekskonfigurationen, die auf spezifische Forschungsziele zugeschnitten sind.
Vorteile unseres 2b-RAD-Services
- Durch erhebliche Flexibilität gekennzeichnet, ist die Anzahl der Tags steuerbar.
- Die Tags weisen konsistente Längen auf, um eine einheitliche Amplifikationseffizienz während der PCR zu gewährleisten.
- Diese Tags zeigen positionsspezifische Eigenschaften, unabhängig von Referenzsequenzen und Assembly-Ergebnissen.
- Neben der Verwendung von co-dominanten Markern wie SNPs können auch dominante Marker eingesetzt werden.
- Genau und erschwinglich
- Flexibler Tag-Nummer
- Konsistente Etikettenlänge
- Hohe Dichte von Markern
- Keine Zwischenreinigungsstufen erforderlich
- Ermöglichung von niedrigem DNA-Eingang und degradierter DNA
- Hochgradig reduzierte 2b-RAD-Bibliotheken erfordern deutlich weniger Sequenzierung für eine genaue Genotypisierung.
Anwendungen von 2b-RAD
- Bin-Kartenkonstruktion und QTL-Standort
- Populationsgenetik Studium
- Bevölkerungsentwicklung Analyse
- Genomweite Assoziationsstudie
- Genomphylogenie
- Genomische Selektion
- Verknüpfungs- und Assoziationsmapping
- Diskriminierung von mikrobiellen Stämmen
- Erkennung somatischer Mutationen
2b-RAD Arbeitsablauf
Der allgemeine Arbeitsablauf für 2b-RAD umfasst die Probenvorbereitung, die Vorbereitung der 2b-RAD-Bibliothek, Hochdurchsatz-Sequenzierung und bioinformatische Analyse. Unser hochqualifiziertes Expertenteam führt das Qualitätsmanagement durch und überwacht jeden Schritt, um zuverlässige und unvoreingenommene Ergebnisse zu gewährleisten. Der Bibliotheksaufbau für 2b-RAD besteht aus vier Hauptphasen (BsaXI-Digestion, Ligation, Amplifikation und Barcoding).

Dienstspezifikationen
Musteranforderungen
|
|
 Klicken |
Sequenzierungsstrategie
|
 |
Bioinformatikanalyse
Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an:
|
Analyse-Pipeline
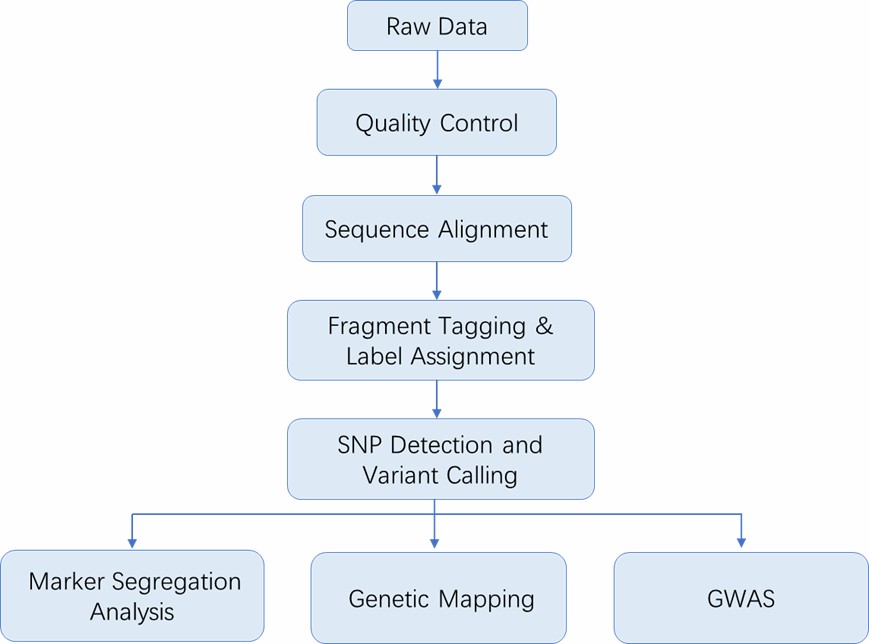
Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details in 2b-RAD für Ihr Schreiben (Anpassung)
CD Genomics freut sich, modernste 2b-RAD-Sequenzierungstechnologie anzubieten, die einen umfassenden Service umfasst, der den gesamten Workflow von der Qualitätskontrolle genomischer DNA bis zur Bestimmung von Genotypen und der Bewertung der Populationsvielfalt abdeckt. Wir laden Sie ein, uns zu kontaktieren, wenn Sie spezifische Bedürfnisse oder Anfragen zu unseren Dienstleistungen haben. Als lizenzierter Anbieter dieser fortschrittlichen Technologie gewährleistet CD Genomics die Einhaltung von Standards und Exzellenz bei der Bereitstellung robuster genomischer Lösungen. Es ist wichtig zu beachten, dass Keygene N.V. die Patente und Patentanmeldungen hält, die das geistige Eigentum im Zusammenhang mit sequenzbasierten Genotypisierungstechnologien schützen.
Demo-Ergebnisse
Teilweise Ergebnisse sind unten aufgeführt:
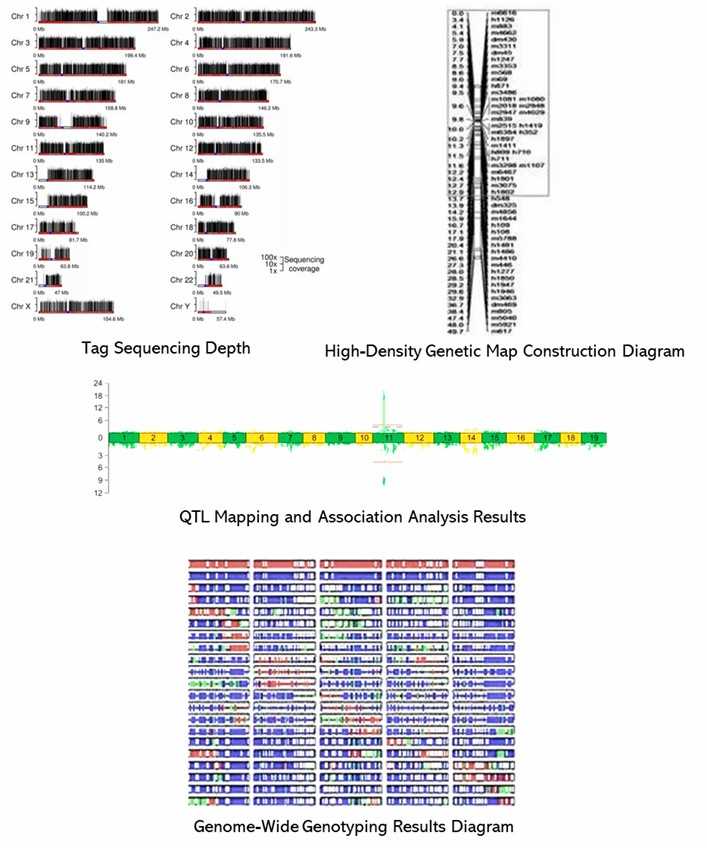
2b-RAD häufig gestellte Fragen (FAQs)
1. Was sind die Vorteile von 2b-RAD?
CD Genomics ist stolz darauf, die fortschrittliche 2b-RAD-Sequenzierungstechnologie anzubieten, die zahlreiche Vorteile gegenüber der ursprünglichen bietet. RAD-seq und GBSDer Vergleich zwischen verschiedenen Methoden der reduzierten Repräsentation bei der Ganzgenomsequenzierung ist in Tabelle 1 dargestellt.
Tabelle 1. Technischer Vergleich zwischen 2b-RAD und anderen NGS-basierten Methoden zur reduzierten Repräsentationsgenotypisierung.
| Technologie | RAD-seq, ddRAD | GBS | 2b-RAD |
|---|---|---|---|
| Bibliotheksbau | Komplex, einschließlich Sonikation, Fragmentauswahl und mehreren Schritten der DNA-Reinigung. | Keine Sonikation und Fragmentauswahl, Endreparatur | Vereinfacht, keine Sonikation und Fragmentauswahl, Endreparatur |
| Fragmentgröße | Nicht einheitlich | Nicht einheitlich | Uniform |
| Tag-Dichte-Anpassung | Schwierig | Möglich, aber nicht getestet | Flexibel |
| Tag-Sequenzierungstiefe | Hochgradig divergente Tiefe zwischen verschiedenen Tags | Hochgradig divergente Tiefe zwischen verschiedenen Tags | Einheitliche Sequierungstiefe zwischen Tags |
| Datenanalyse | Falsch-positive Ergebnisse verursacht durch repetitive Sequenzen | Falsch-positiv verursacht durch repetitive Sequenzen | Entfernen Sie die Ablenkung durch sich wiederholende Sequenzen, indem Sie iML verwenden. |
2. Was sind die Nachteile von 2b-RAD?
Obwohl 2b-RAD eine leistungsstarke Methode für Hochdurchsatz ist Genotypisierung, es hat auch einige Nachteile. 2b-RAD funktioniert nur bei diploiden Arten. Darüber hinaus sind die kurzen Tags möglicherweise nicht lang genug für die Lokusdiskriminierung in komplexen Genomen.
Wie wird die 2b-RAD-Bibliothek vorbereitet?
Der Bibliotheksaufbau für 2b-RAD besteht aus vier Hauptphasen (BsaXI-Digestion, Ligation, Amplifikation und Barcoding). Nach der DNA-Digestion durch BsaXI werden Adapter über kohäsive Enden an die Fragmente ligiert, und spezifische Barcodes werden durch PCR-Amplifikation unter Verwendung von degenerierten Linkern in jede Probe integriert. Anschließend werden die Einzel-Tag-Konstrukte unter Verwendung modifizierter Adapter und biotin-markierter Primer hergestellt, die weiter durch SapI verdaut werden können, um unterschiedliche kohäsive Enden zu erzeugen, die dann in einer vordefinierten Reihenfolge ligiert werden, um fünf verkettete Tags zu produzieren. Die Proben werden dann zusammengeführt und mit Illumina-Technologie sequenziert.
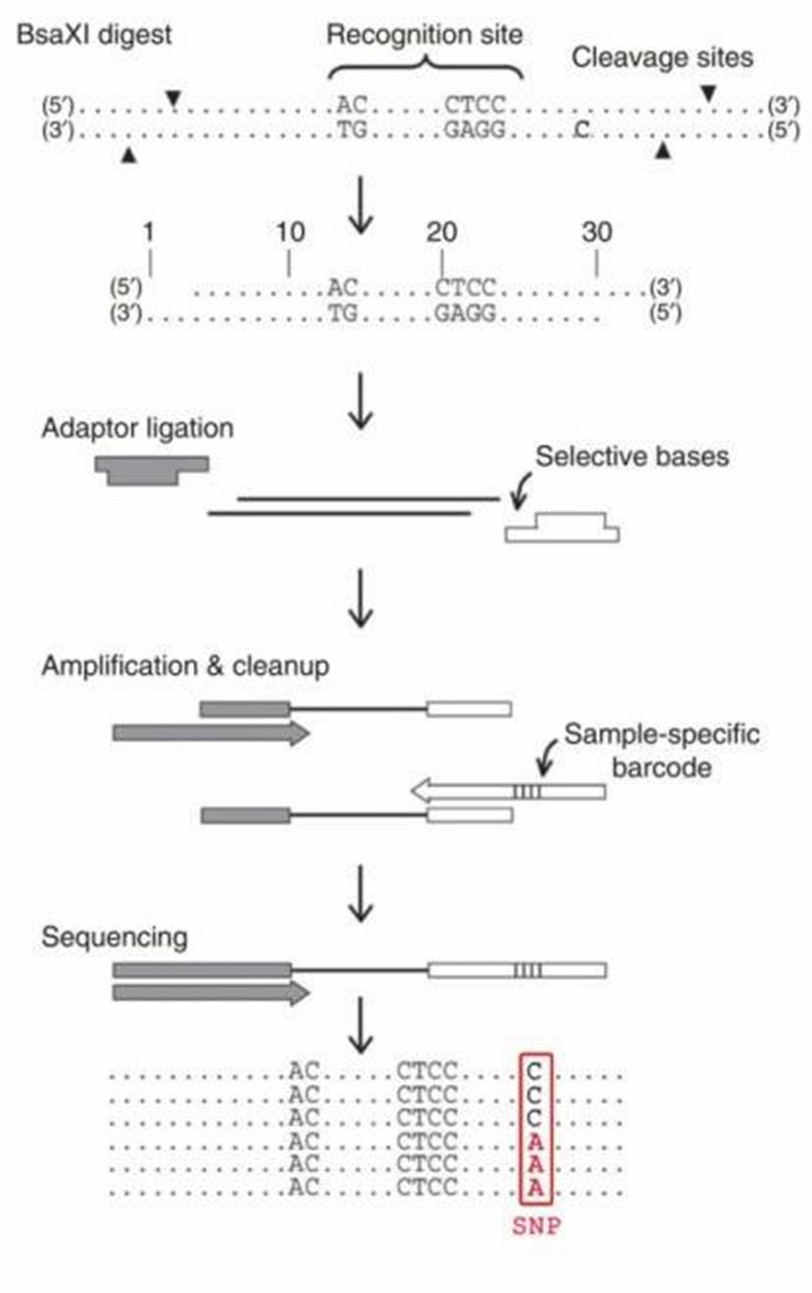 Abbildung 1. Schematische Übersicht des 2b-RAD-Verfahrens.
Abbildung 1. Schematische Übersicht des 2b-RAD-Verfahrens.
4. Kann 2b-RAD SNPs in repetitiven Regionen nachweisen?
2b-RAD kann SNPs in repetitiven Regionen, die nur eine geringe Anzahl von Restriktionsstellen enthalten, möglicherweise nicht erkennen.
5. Wie viele Individuen sind für den Bau einer Verknüpfungskarte erforderlich?
Mindestens mehr als 100 Personen sind erforderlich für den Bau. genetische Verknüpfungskarte.
6. Wie viele Proben sind für natürliche Populationen erforderlich?
Natürliche Populationen haben oft mehr SNPs. Die Anzahl der Proben hängt vom interessierenden Phänotyp ab und reicht von mehreren Dutzend bis zu mehreren Hundert. Je mehr Proben Sie einreichen, desto genauer sind die Genotypisierungsergebnisse.
7. Was ist die Anforderung an Arten?
Diploide mit oder ohne Referenzgenom sind für 2b-RAD geeignet. Polyploide sollten jedoch besser ein Referenzgenom haben, und es bestehen weiterhin Risiken.
2b-RAD Fallstudien
Eine hochauflösende genetische Verknüpfungskarte und feine QTL-Kartierung für wachstumsbezogene Merkmale und Geschlecht beim Yangtze-River-KarpfenCyprinus carpio haematopterus)
Journal: BMC Genomik
Veröffentlicht: 02. April 2018
Hintergrund
Die Autoren konstruierten eine hochauflösende genetische Verknüpfungskarte durch die Nutzung von 7820 2b-RAD und 295 Mikrosatellitenmarkern in einer F2-Familie des Karpfens aus dem Yangtze-FlussC. c. Haematopterus). Sie kartierten eine Reihe von vielversprechenden und signifikanten QTLs für wachstumsbezogene Merkmale und Geschlecht auf diesem Verknüpfungskarten. Die genetische Karte sowie diese aus QTL abgeleiteten Kandidatengene und Marker sind nützlich für weitere Studien zum Karpfen des Yangtze.
Materialien & Methoden
Probenvorbereitung
- Gemeiner Karpfen
- DNA-Extraktion
Sequenzierung
- 2b-RAD-Sequenzierung
- De novo Genotypisierung
- Mikrosatelliten-Genotypisierung
- Verknüpfungskartenkonstruktion
- Vergleichende Genomanalyse
- QTL-Analyse
Ergebnisse
1. Erstellung der hochauflösenden Verknüpfungskarte
Insgesamt wurden 8115 Marker (7820 2b-RAD-Marker und 295 SSRs) in 50 LGs gruppiert, was mit der haploiden Chromosomenzahl übereinstimmte, indem die Software JoinMap 4.1 verwendet wurde.
 Abbildung 1. Der geschlechtsdurchschnittliche genetische Verknüpfungsplan des Karpfens im Jangtse. C.c. Haematopterus konstruiert auf der Grundlage von 2b-RAD- und Mikrosatellitenmarkern.
Abbildung 1. Der geschlechtsdurchschnittliche genetische Verknüpfungsplan des Karpfens im Jangtse. C.c. Haematopterus konstruiert auf der Grundlage von 2b-RAD- und Mikrosatellitenmarkern.
2. Vergleichende Genomkartierung
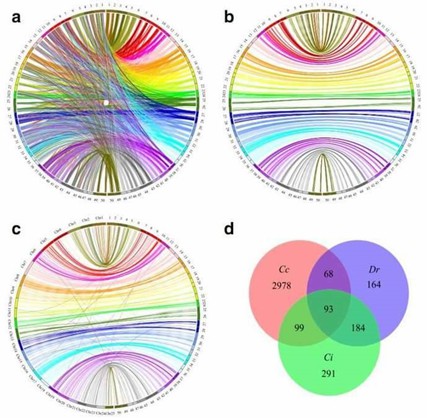 Abbildung 2. Circos-Diagramm, das syntenische Beziehungen zwischen C. c. Haematopterus (rechts) und (a und b) C. c. Carpio (links) und Danio rerio und (d) Venn-Diagramme, die Überlappungen zwischen einzigartig ausgerichteten Markern beschreiben, die auf das Genom von C. c. carpio (Cc), D. rerio (Dr) und C. Idellus (Ci).
Abbildung 2. Circos-Diagramm, das syntenische Beziehungen zwischen C. c. Haematopterus (rechts) und (a und b) C. c. Carpio (links) und Danio rerio und (d) Venn-Diagramme, die Überlappungen zwischen einzigartig ausgerichteten Markern beschreiben, die auf das Genom von C. c. carpio (Cc), D. rerio (Dr) und C. Idellus (Ci).
3. Feine QTL-Kartierung für wachstumsbezogene Merkmale und Geschlecht.
Insgesamt wurden 21 QTLs, die mit wachstumsbezogenen Merkmalen assoziiert sind, auf 12 LGs identifiziert, darunter zwei genomweit signifikante QTLs und 19 chromosomenweit signifikante QTLs.
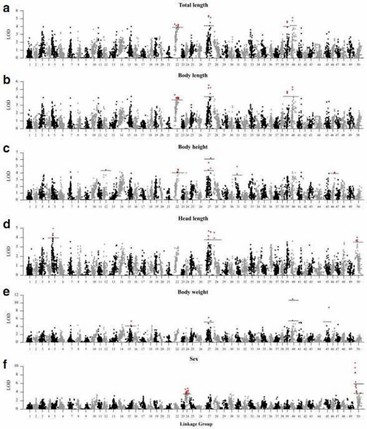 Abbildung 3. Eine Genomscan von LOD-Profilen für (a) Gesamtlänge, (b) Körperlänge, (c) Körperhöhe, (d) Kopflänge, (e) Körpergewicht und (f) Geschlecht in C. c. HämatopterusDie gestrichelten und durchgezogenen Linien zeigen die chromosomenweiten und genomweiten Signifikanzschwellen an.
Abbildung 3. Eine Genomscan von LOD-Profilen für (a) Gesamtlänge, (b) Körperlänge, (c) Körperhöhe, (d) Kopflänge, (e) Körpergewicht und (f) Geschlecht in C. c. HämatopterusDie gestrichelten und durchgezogenen Linien zeigen die chromosomenweiten und genomweiten Signifikanzschwellen an.
4. Potenzielle Kandidatengene für das Wachstum
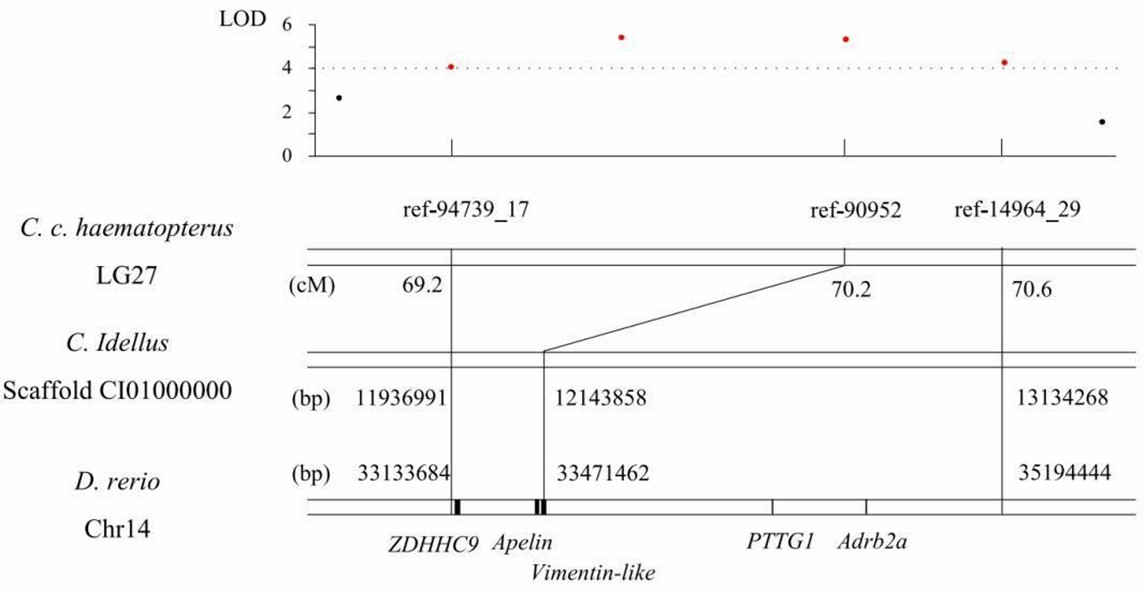 Abbildung 4. Der QTL-Bereich für Wachstumsmerkmale auf LG27 von C. c. Hasematopterus und seine homologe Region in Genomen von D. rerio und C. idellus.
Abbildung 4. Der QTL-Bereich für Wachstumsmerkmale auf LG27 von C. c. Hasematopterus und seine homologe Region in Genomen von D. rerio und C. idellus.
Referenz
- Feng X, Yu X, Fu B, u. a.Eine hochauflösende genetische Verknüpfungskarte und feine Kartierung von QTL für wachstumsbezogene Merkmale und Geschlecht beim Karpfen aus dem Yangtze.Cyprinus carpio haematopterus). BMC Genomics, 2018, 19(1): 230.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Treiber der genomischen Vielfalt und phänotypischen Entwicklung in den frühen Phasen der Domestikation von Hermetia illucens
Zeitschrift: Insekten-Molekularbiologie
Jahr: 2024
Die Restriktions-Modifikationssysteme von Clostridium carboxidivorans P7
Journal: Mikroorganismen
Jahr: 2023
Im Land der Blinden: Außergewöhnliche subterranne Spezialisierung von kryptischen troglobitischen Spinnen der Gattung Tegenaria (Araneae: Agelenidae) in Israel
Zeitschrift: Molekulare Phylogenetik und Evolution
Jahr: 2023
Genetische Modifikatoren des oralen Nikotinkonsums bei Chrna5-Nullmutantenmäusen
Zeitschrift: Front. Psychiatrie
Jahr: 2021
Eine hochdichte genetische Verknüpfungskarte und QTL-Identifizierung für Wachstumsmerkmale bei Dunkelkob (Argyrosomus japonicus)
Zeitschrift: Aquakultur
Jahr: 2024
Genomische und chemische Beweise für lokale Anpassung an die Resistenz gegenüber verschiedenen Herbivoren in Datura stramonium
Journal: Evolution
Jahr: 2020
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


