
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
ATAC-Seq
CD Genomics kann jetzt Assays für transposasezugängliche Chromatin mit Hochdurchsatz-Sequenzierung (ATAC-seq) anbieten, eine Methode zur Kartierung der Chromatinzugänglichkeit im gesamten Genom. Die Methode ist eine schnelle und empfindliche Alternative zu DNase-seq (Sequenzierung von DNase I-hypersensitiven Stellen) oder MNase-seq (Sequenzierung von mikrokokkal-nukleaseempfindlichen Stellen). Mit unserem Service können Sie genomweite Profile von offenen und zugänglichen Chromatinregionen erkennen, die auf aktive regulatorische Regionen hinweisen.
Die Einführung von ATAC-Seq
Das eukaryotische Genom ist stark verpackt, um in den sehr begrenzten nukleären Raum zu passen. Infolgedessen wird der Zugang zu genomischen Informationen streng reguliert, basierend auf dem Zellzustand. Welche Regionen des Genoms zugänglich sind, verrät viel über den Zustand der Zelle. ATAC-seq ist eine Technik, um zugängliche Chromatinregionen zu lokalisieren.
Das Akronym ATAC-seq steht für Assay for Transposase Accessible Chromatin using sequencing, eine Technik, die die Zugänglichkeit von Chromatin durch Transposase mittels Sequenzierung untersucht. Dieser Ansatz beinhaltet den Schnitt offener Chromatinregionen in spezifischen zeitlichen und räumlichen Kontexten durch Transposase, wodurch regulatorische Sequenzen aktiv transkribierter Gene im Genom zu diesem spezifischen Zeitpunkt erfasst werden. Diese Methode erfordert nur eine kleine Anzahl von Zellen und liefert Echtzeitinformationen über die regulatorischen Sequenzen, die die genomweite Aktivität steuern. Zu den Anwendungen gehören die Analyse der Bindung von Transkriptionsfaktoren, die Positionierung von Nukleosomen und die Verteilung regulatorischer Elemente, was enorme Perspektiven im Bereich der epigenetischen Forschung bietet.
ATAC-seq ist eine innovative Technik in der epigenetischen Forschung, die Tn5-Transposase verwendet, um offene Chromatinstellen zu schneiden und zu kennzeichnen, was eine effiziente Sequenzierung von Chromatinzugänglichkeitskarten ermöglicht. Die Tn5-Transposase bindet zufällig und schneidet DNA in offenen Chromatinregionen, während sie gleichzeitig Adaptersequenzen an den Schnittstellen einfügt. Durch die Einführung des Transposase-Komplexes, der bekannte DNA-Sequenzmarkierungen (d.h. Tn5-Transposase mit roten und grünen Sequenzmarkierungen) trägt, in den Zellkern zur Ko-Inkubation und anschließender PCR-Amplifikation unter Verwendung der bekannten Sequenzmarkierungen kann eine Bibliothek erstellt werden, und die Sequenzierung kann Informationen über offene Chromatinregionen liefern.
Vorteile von ATAC-Seq
- Gewinnen Sie mechanistische Einblicke in die Genregulation, die zelluläre Reaktion auf Behandlung oder Krankheit.
- Identifizieren Sie, welche Transkriptionsfaktoren das Zellschicksal, Krankheiten oder Reaktionen steuern.
- Begrenzte Patientenproben
- Geringe Anforderungen an die Menge der biologischen Probe, und das gesamte Protokoll benötigt insgesamt 3 Stunden.
Anwendungen von ATAC-Seq
- Nukleosomenpositionierung
- Identifizierung von Schlüsseltranskriptionsfaktoren
- Erkennung von Promotorregionen, potenziellen Enhancern oder Silencern
- Integration von Multi-Omics-Daten
- Kartierung der Chromatinzugänglichkeit
- Identifizierung von durch Transkriptionsfaktoren regulierten Zielgenen
ATAC-Seq Arbeitsablauf
Der zentrale Bestandteil des ATAC-seq-Verfahrens ist die Wirkung der Transposase Tn5 auf die genomische DNA der Probe. Transposasen sind Enzyme, die die Bewegung von Transposons zu anderen Teilen des Genoms katalysieren. Während natürlich vorkommende Transposasen ein niedriges Aktivitätsniveau aufweisen, verwendet ATAC-seq eine mutierte hyperaktive Transposase. Die hohe Aktivität ermöglicht ein äußerst effizientes Schneiden von exponierter DNA und die gleichzeitige Ligation spezifischer Sequenzen, die als Adapter bezeichnet werden. Adapter-ligierte DNA-Fragmente werden dann isoliert, durch PCR amplifiziert und verwendet für Next-Generation-SequenzierungDie experimentelle Pipeline von ATAC-seq wird in Abbildung 1 dargestellt.
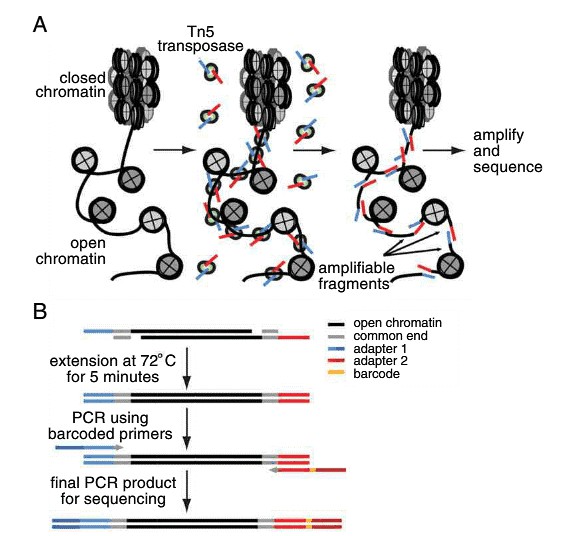 Abbildung 1. Schematischer Arbeitsablauf des ATAC-seq-Prozesses.
Abbildung 1. Schematischer Arbeitsablauf des ATAC-seq-Prozesses.

Dienstspezifikation
Musteranforderungen
|
|
 Klicken |
Sequenzierungsstrategien
|
 |
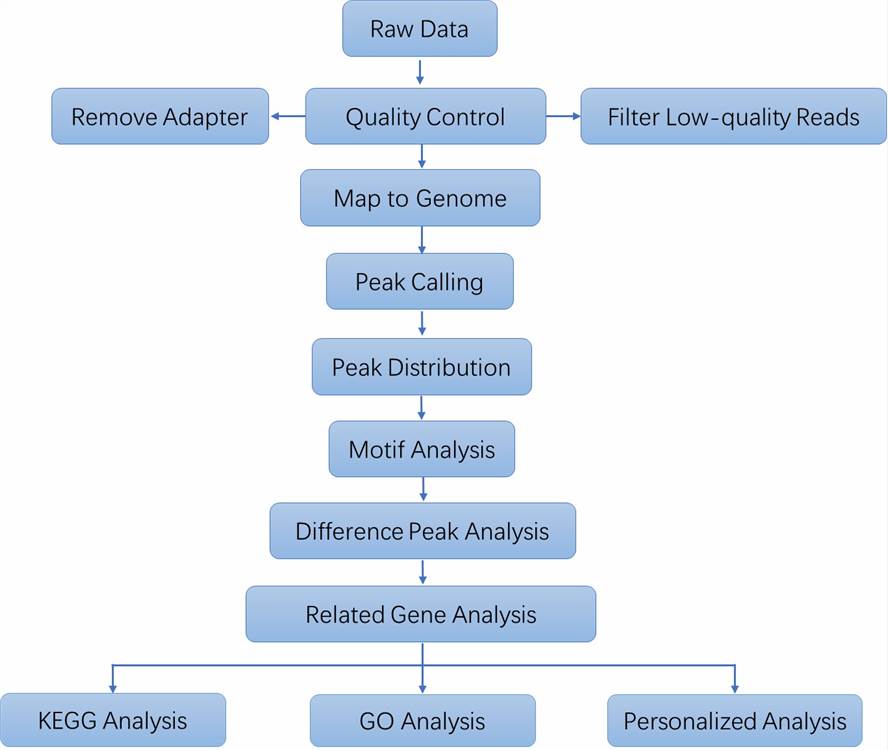
Datenanalyse Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an:
|
Analyse-Pipeline
Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details in ATAC-Seq für Ihr Schreiben (Anpassung)
CD Genomics versichert, dass wir einen hochwertigen Service bieten werden, und unser professionelles Team wird Sie nicht enttäuschen. Mit mehr als 10 Jahren Erfahrung in der Unterstützung von Forschern weltweit im Bereich NGS denke ich, dass wir das sein könnten. Zögern Sie bitte nicht, uns zu kontaktieren, wenn Sie Fragen zu unserem Service haben.
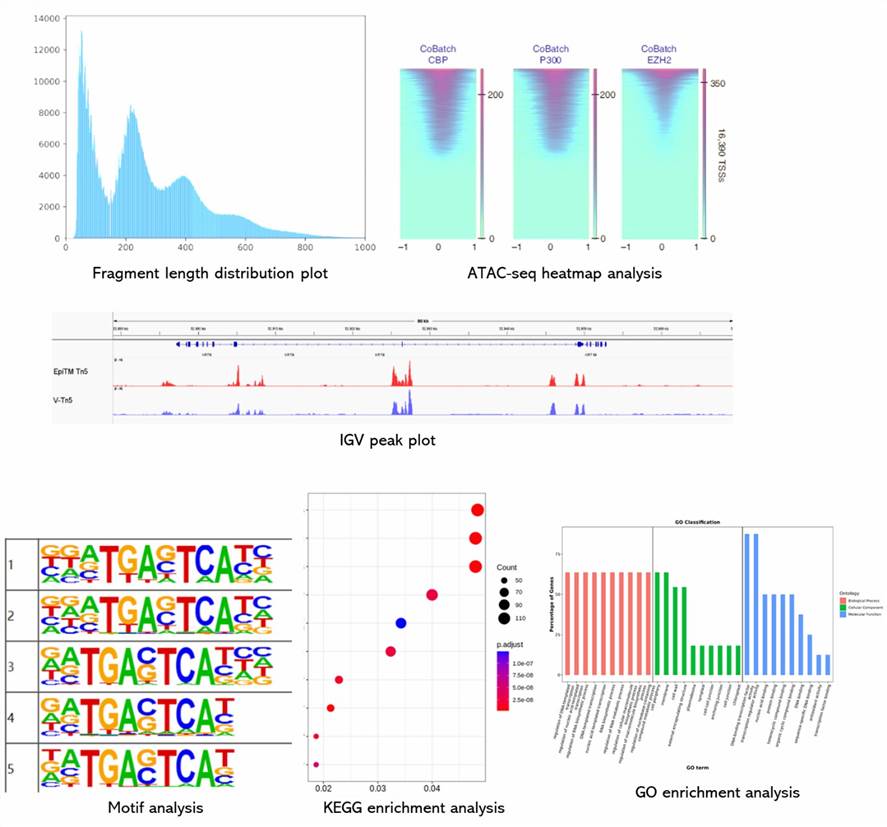
Demo-Ergebnisse
Teilweise Ergebnisse sind unten aufgeführt:
ATAC-Seq häufig gestellte Fragen (FAQs)
1. Welche Art von Kontrollen sind für ATAC-Seq-Experimente notwendig?
Im Bereich der ATAC-Seq-Experimente ist die Einbeziehung verschiedener Kontrollen unerlässlich, um Präzision und Zuverlässigkeit aufrechtzuerhalten. Die Verwendung einer Input-DNA-Kontrolle dient dazu, potenzielle Verzerrungen in der DNA-Vorbereitung und den Sequenzierungsprozessen zu normalisieren. Technische Replikate spielen eine entscheidende Rolle bei der Bestätigung der Reproduzierbarkeit und Zuverlässigkeit der Ergebnisse. Die Einbeziehung positiver Kontrollen, die etablierte offene Chromatinregionen repräsentieren, fungiert als Mechanismus zur Validierung des Protokolls. Auf der anderen Seite helfen negative Kontrollen, die erwartete geschlossene Chromatinregionen anzeigen, bei der Erkennung und Minderung von Hintergrundgeräuschen.
2. Wie schneidet ATAC-Seq im Vergleich zu anderen Chromatin-Zugänglichkeitsassays wie DNase-Seq und FAIRE-Seq ab?
DNase-Seq, ATAC-Seq und FAIRE-Seq sind Techniken, die verwendet werden, um offene Chromatinregionen zu untersuchen. DNase-Seq nutzt die DNase I Endonuklease, um zugängliche Chromatinregionen zu identifizieren, FAIRE-Seq umfasst Sonikation gefolgt von Phenol-Chloroform-Anreicherung, und ATAC-Seq verwendet Tn5 Transposase zur Anreicherung und Amplifikation. Die hohe Aktivität der Tn5 Transposase macht ATAC-Seq zu einer einfachen, effizienten Methode, die nur 500-50.000 Zellen benötigt. Die Sensitivität und Spezifität von ATAC-Seq sind vergleichbar mit DNase-Seq und überlegen gegenüber FAIRE-Seq.
3. Welche Techniken werden häufig mit ATAC-seq für die Forschung kombiniert?
ATAC-seq + RNA-SeqIm Allgemeinen wird RNA-seq vor ATAC-seq durchgeführt, um differentiell exprimierte Gene und angereicherte Signalwege zu identifizieren, die Korrelationen nahelegen. Um zu bestimmen, welche Faktoren diese Zielgene regulieren, kann die Motivanalyse über ATAC-seq potenzielle regulatorische Elemente identifizieren. Eine anschließende Validierung kann durch zusätzliche Experimente oder ChIP-seq erfolgen.
ATAC-seq + Hi-CFür Studien, die die Auswirkungen von höherordentlichen Chromatinstrukturen auf biologische Prozesse untersuchen, wird ATAC-seq häufig zusammen mit Hi-C verwendet. Hi-C liefert Informationen über höherordentliche Strukturen wie Kompartimente A/B, TADs und Schleifen, die Korrelationen anzeigen. ATAC-seq kann zudem Promotoren, Enhancer und andere Elemente identifizieren und aufzeigen, wie diese Strukturen die Genexpression beeinflussen.
ATAC-seq + HistonmodifikationenWährend ATAC-seq die Zugänglichkeit spezifischer Stellen und potenzieller Transkriptionsfaktor-Bindungen vorhersagen kann, zeigt es nicht, ob diese Faktoren die Genexpression fördern oder hemmen. Die Kombination von ATAC-seq mit Daten zu Histonmodifikationen (z. B. H3K27ac für Aktivierung oder H3K27me3 für Repression) kann ein umfassenderes Verständnis bieten und die Daten robuster und zuverlässiger machen.
ATAC-Seq Fallstudien
Multiomische Analysen der Auswirkungen von LED-weißem Licht auf die Reifung von Aprikosenfrüchten
Zeitschrift: Zeitschrift für Fortgeschrittene Forschung
Impact-Faktor: 12,822
Veröffentlicht: Online verfügbar am 9. Januar 2024
Hintergrund
AprikosePrunus armeniaca L.., die in Zentralasien heimisch ist und in Regionen wie dem Mittelmeerraum und China weit verbreitet angebaut wird, ist aufgrund ihrer Farbe, ihres Geschmacks und ihrer Nährstoffe beliebt, hat jedoch eine kurze Lagerdauer von 3-5 Tagen. Der Reifungs- und Alterungsprozess, der durch Ethylen reguliert wird, umfasst komplexe physiologische Veränderungen. Trotz umfangreicher Forschung sind die Veränderungen der Metaboliten und die Regulierungsmechanismen während der Nachernte-Lagerung nicht vollständig verstanden. Lichtemittierende Dioden (LEDs) bieten eine umweltfreundliche Methode, um die Haltbarkeit zu verlängern, indem sie die Alterung verzögern und die Qualität erhalten. Diese Studie untersucht die Auswirkungen von LED-weißem Licht auf die Qualität von Aprikosenfrüchten, Transkriptom, Metabolom und Chromatinzugänglichkeit, mit dem Ziel, Lagerungsmethoden zu verbessern und die Haltbarkeit zu verlängern.
Materialien & Methoden
- Probenvorbereitung
- Reife Aprikosenfrüchte
- 600 unbeschädigte und fäulnisfreie Früchte
- RNA-Extraktion und -Nachweis
- Zellensammlung
- Bibliothekskonstruktion
- Transkriptom-Sequenzierung
- ATAC-Sequenzierung
- Transpositionsreaktion und Reinigung
- Screening und Analyse von differentiell exprimierten Genen
- Anreicherungsanalyse
- Analyse differenziell zugänglicher Regionen
- Genfunktionsanalyse
Ergebnisse
In dieser Studie wurde die Transkriptomik von Früchten genutzt, um PCC als Index zur Bewertung der biologischen Replikatkorrelation zu verwenden, wobei hohe Werte (>0,9) festgestellt wurden, die auf eine starke Ähnlichkeit hinweisen (Abb. 1F). Die Analyse ergab unterschiedliche Muster der Genexpression in den Vergleichen: CK-0 d vs. CK-8 d (6.256 DEGs), CK-0 d vs. LED-8 d (8.110 DEGs) und CK-8 d vs. LED-8 d (1.792 DEGs), mit variierenden Überlappungen (Abb. 1I). Die KEGG- und GO-Anreicherung hob Wege wie die Biosynthese sekundärer Metaboliten und die regulatorischen Rollen von DEGs in Bezug auf Geschmack, Textur, Farbe, Ethylenbiosynthese, Transkriptionsfaktoren und oxidative Prozesse hervor. LED-Licht regulierte 56 DEGs bei der Fruchtreifung, was Auswirkungen auf Stoffwechselwege und transkriptionale Aktivitäten hatte.
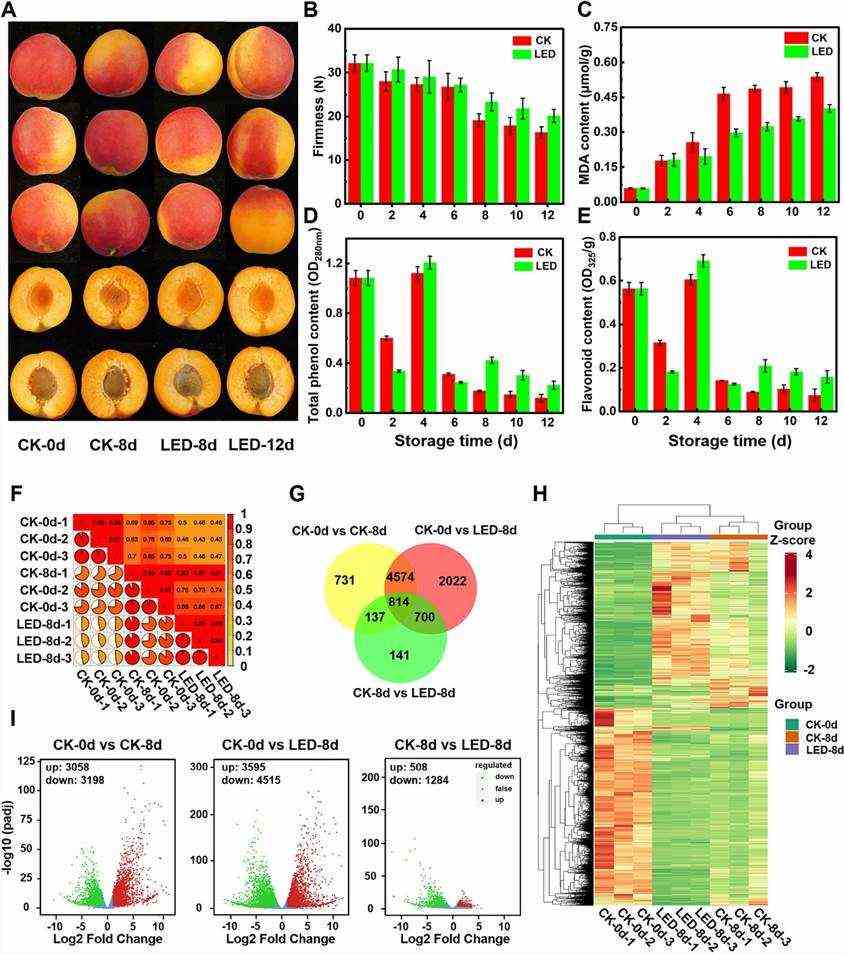 Abb. 1. Auswirkungen der LED-Behandlung auf die physiologische Qualität und Transkription von Aprikosenfrüchten.
Abb. 1. Auswirkungen der LED-Behandlung auf die physiologische Qualität und Transkription von Aprikosenfrüchten.
Kombinierte transkriptomische und metabolomische Analysen zeigten, dass unter LED-Bestrahlung UDP-Zucker-Pyrophosphorylase (USP hochreguliert wurde, was die Synthese von D-Glucuronat erhöhte, während Inositol-Oxygenase (MIOX) und Aldehyddehydrogenase (ALDH) herunterreguliert wurden, was die Ansammlung von Myo-Inositol verringerte. Die LED-Behandlung verringerte auch die Ansammlung von L-Dehydroascorbinsäure, indem sie die Aktivitäten von L-Ascorbat-Peroxidase (APX) und L-Ascorbat-Oxidase (AO) beeinflusste. Darüber hinaus führten LED-induzierte Erhöhungen der Cinnamoyl-CoA-Spiegel zur Hochregulierung der Chalcon-Synthase (CHS) und der Phlorizin-Synthase (PTG1), was den Flavonoidstoffwechsel veränderte und die Anthocyanin-Biosynthese durch die Hochregulierung der Dihydroflavonol-4-Reduktase (DFR) förderte.
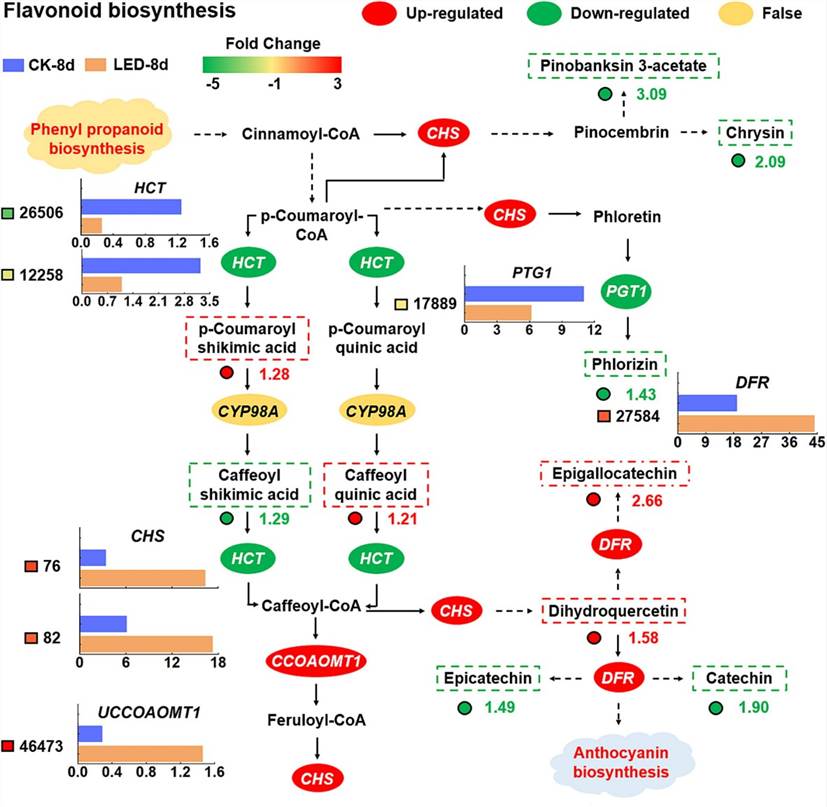 Abb. 2. Der Stoffwechselweg der Flavonoidbiosynthese wurde durch die Kombination von Transkriptom- und Metabolomdaten analysiert.
Abb. 2. Der Stoffwechselweg der Flavonoidbiosynthese wurde durch die Kombination von Transkriptom- und Metabolomdaten analysiert.
Die ATAC-seq-Analyse von Aprikosenfrüchten nach der LED-Behandlung ergab 616,85 Millionen saubere Reads mit hohen Q30-Basenanteilen, was auf eine robuste Datenqualität hinweist. Die Korrelationsanalyse zeigte eine starke Reproduzierbarkeit unter biologischen Replikaten innerhalb jeder Gruppe. Die Differenzialanalyse identifizierte 4492, 1769 und 123 unterschiedlich zugängliche Regionen (DARs) in CK-0 d vs. CK-8 d, CK-0 d vs. LED-8 d und CK-8 d vs. LED-8 d, jeweils. Die Motiv-Analyse hob 76 Transkriptionsfaktoren (TFs hervor, darunter ethylen-responsive Faktoren (ERF2, ERF9, ERF069, ERF104 und MYB124), die an der Regulierung von DARs und Zielgenen beteiligt sind. Diese Ergebnisse unterstreichen die durch LEDs induzierten Veränderungen in der Chromatin-Zugänglichkeit und der TF-Regulation, die die Genexpression und die Fruchtentwicklung bei Aprikosen beeinflussen.
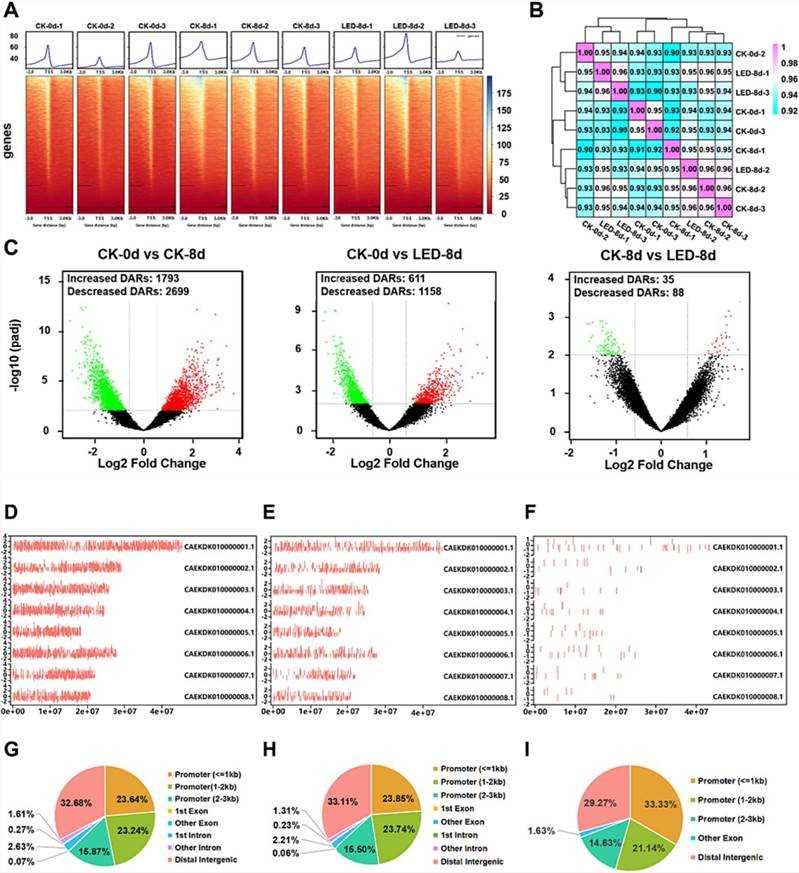 Abb. 3. Übersicht der ATAC-seq-Daten zur Reifung von Aprikosenfrüchten.
Abb. 3. Übersicht der ATAC-seq-Daten zur Reifung von Aprikosenfrüchten.
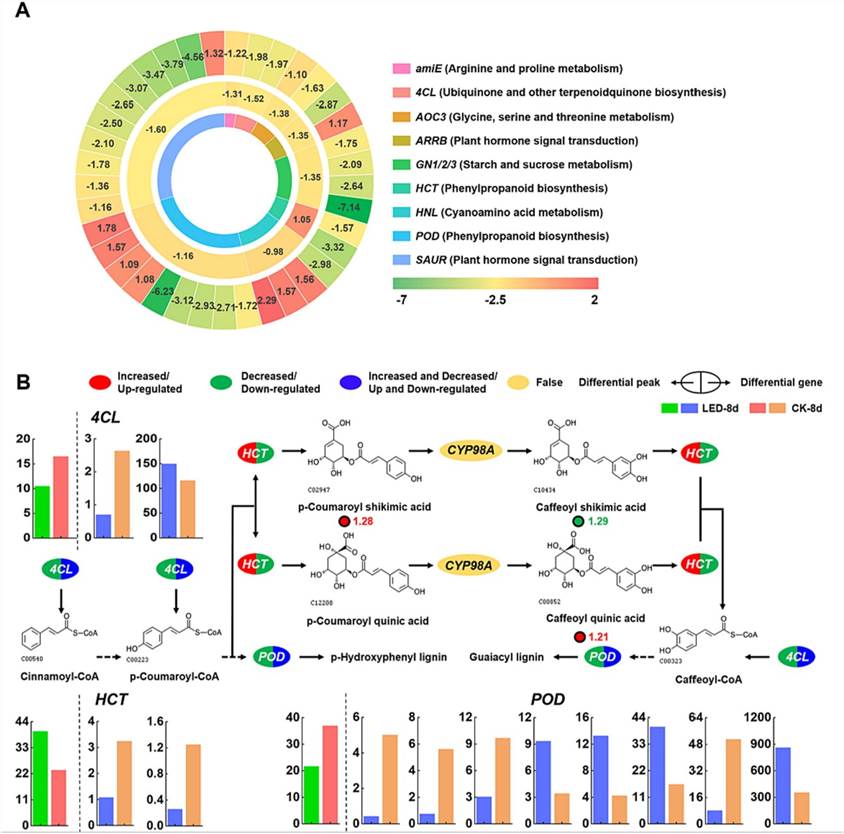 Abb. 4. (A) Differenzielle Genexpression und Heatmap der offenen Chromatinregionen. (B) Der Stoffwechselweg der Phenylpropanoid-Biosynthese wurde durch die Kombination von Transkriptom-, Metabolom- und ATAC-seq-Daten analysiert.
Abb. 4. (A) Differenzielle Genexpression und Heatmap der offenen Chromatinregionen. (B) Der Stoffwechselweg der Phenylpropanoid-Biosynthese wurde durch die Kombination von Transkriptom-, Metabolom- und ATAC-seq-Daten analysiert.
In mit LED-behandelten Aprikosenfrüchten zeigten integrierte Transkriptom- und ATAC-seq-Analysen Gene mit veränderter Chromatinzugänglichkeit und Expressionsniveaus. HCT wies eine reduzierte Chromatinzugänglichkeit und eine verringerte Genexpression auf, während POD und 4CL variable Veränderungen in der Zugänglichkeit zeigten, gepaart mit gemischter Hoch- und Herunterregulierung der Genexpression.
Die metabolomische Analyse hob die Phenylpropanoid-Biosynthese hervor, die durch die LED-Behandlung erheblich beeinflusst wurde. Veränderungen im HCT-Expression beeinflussten die Ansammlung spezifischer Metaboliten wie p-Coumaroyl-Shikimisäure und Caffeoyl-Chinasäure. POD und 4CL, mit veränderter Chromatinzugänglichkeit und variierter Genexpression, spielten eine Rolle in den Ligninsynthesewegen, die entscheidend für die Erhaltung der Qualität von Aprikosenfrüchten während der Lagerung sind.
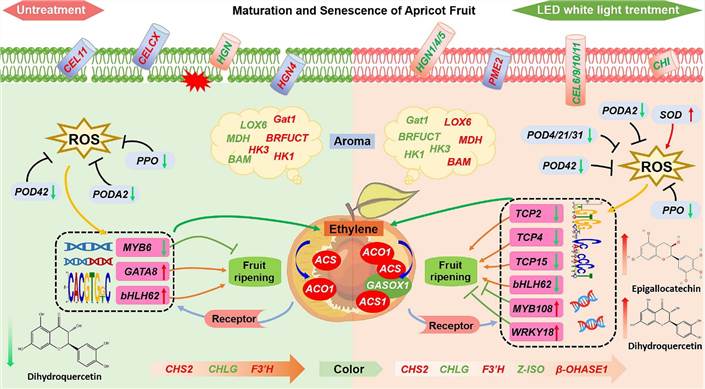 Abb. 5 Modell des Regulationsnetzwerks von LED-Licht während der Reifung von geernteten Aprikosenfrüchten.
Abb. 5 Modell des Regulationsnetzwerks von LED-Licht während der Reifung von geernteten Aprikosenfrüchten.
Fazit
Die LED-Behandlung veränderte die Stoffwechselwege für die Ascorbinsäure- und Uronatmetabolismus, die Flavonoidbiosynthese und die Phenylpropanoidbiosynthese in Aprikosenfrüchten und beeinflusste die Genexpression, die mit der Signalübertragung von Pflanzenhormonen, der Fruchtqualität und der antioxidativen Aktivität verbunden ist. Diese Forschung hebt das Potenzial von LEDs hervor, die Lagerung und Haltbarkeit von Obst und Gemüse zu verlängern.
Referenz:
- Bai C, Zheng Y, Watkins CB, u. a.Multiomik-Analysen der Auswirkungen von LED-weißem Licht auf die Reifung von Aprikosenfrüchten. Zeitschrift für fortgeschrittene Forschung. 2024.
Verwandte Publikationen
Hier sind einige Veröffentlichungen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Die Spaltung von Phagen-DNA durch Restriktionsendonukleasen ermöglicht die Wiederbelebung aus der durch Cas13 induzierten bakteriellen Dormanz.
Zeitschrift: Nature Mikrobiologie
Jahr: 2023
IL-4 fördert die Erschöpfung von CD8.+ CART-Zellen
Zeitschrift: Nature Communications
Jahr: 2024
Hohe-Fett-Diäten während der Schwangerschaft führen zu Veränderungen der DNA-Methylierung und Proteinexpression im Pankreasgewebe der Nachkommen: Ein Multi-Omics-Ansatz
Internationale Zeitschrift für Molekulare Wissenschaften
Jahr: 2024
KMT2A assoziiert mit dem PHF5A-PHF14-HMG20A-RAI1 Subkomplex in Stammzellen des Pankreaskarzinoms und reguliert epigenetisch deren Eigenschaften.
Zeitschrift: Nature Communications
Jahr: 2023
Krebsassoziierte DNA-Hypermethylierung von Polycomb-Zielen erfordert die doppelte Erkennung von Histon H2AK119-Ubiquitinierung und der sauren Tasche des Nukleosoms durch DNMT3A.
Journal: Wissenschaftliche Fortschritte
Jahr: 2024
Genomisches Imprinting-ähnliches monoalleles väterliches Ausdrucksmuster bestimmt das Geschlecht von Kanalkatzenfischen.
Journal: Wissenschaftliche Fortschritte
Jahr: 2022
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


