
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben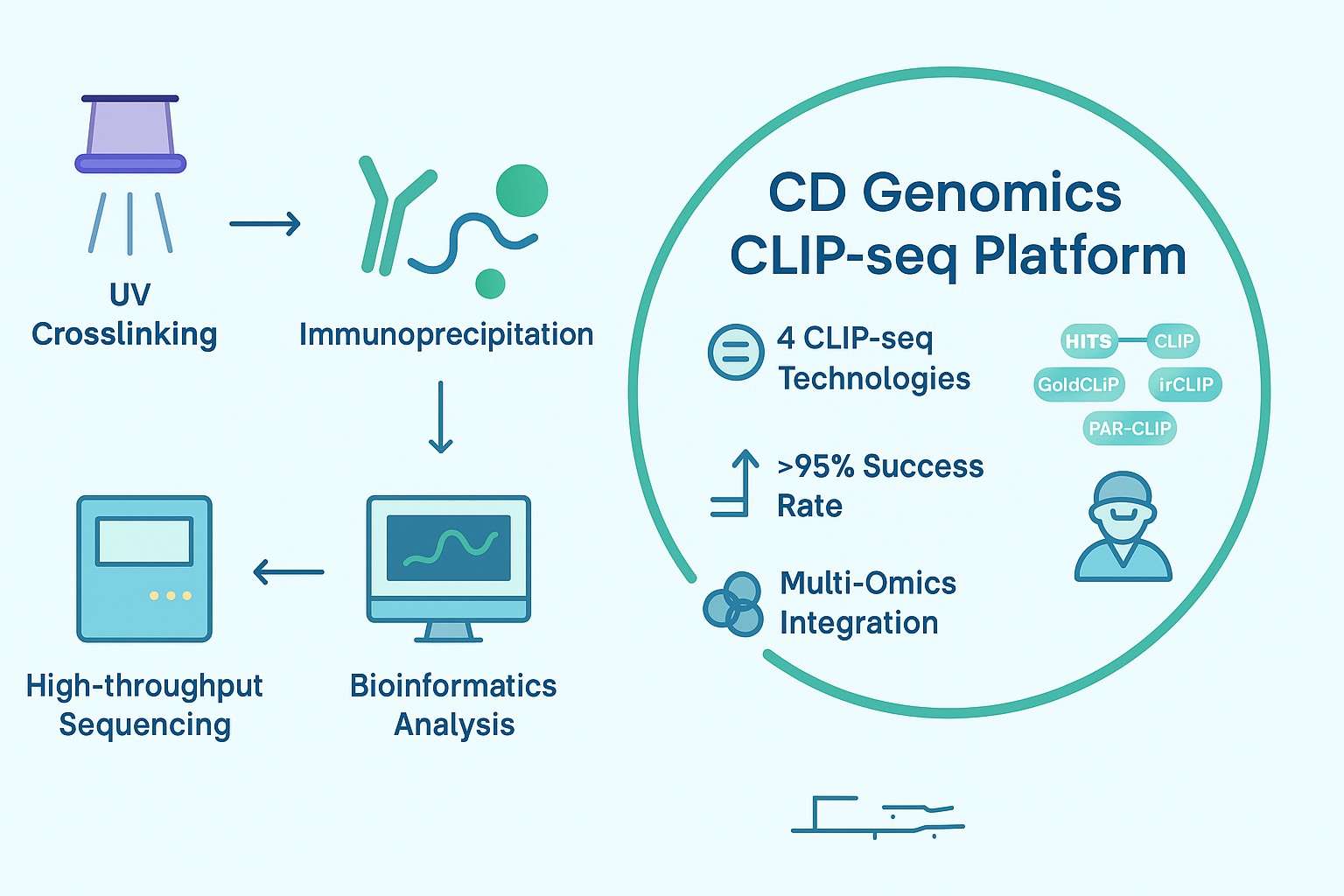
Was ist CLIP-seq?
Wissenschaftler möchten oft wissen, wie Proteine innerhalb von Zellen mit RNA interagieren. Diese Interaktionen sind wichtig, da sie helfen, zu steuern, wie Gene funktionieren und wie sich Zellen verhalten. CLIP-seq, das für "Kreuzvernetzung und Immunpräzipitation gefolgt von Sequenzierung" steht, ist eine Methode, die Forschern hilft, genau herauszufinden, wo Proteine an RNA binden.
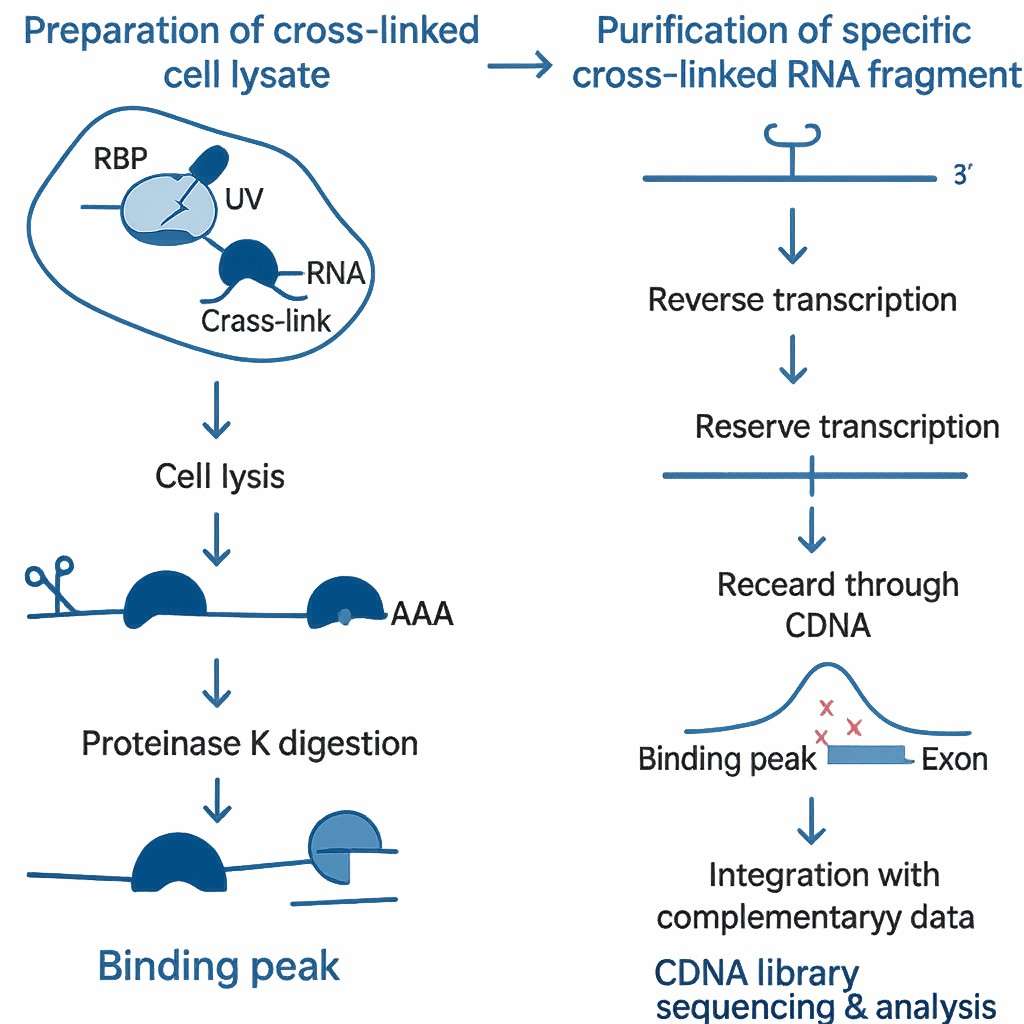
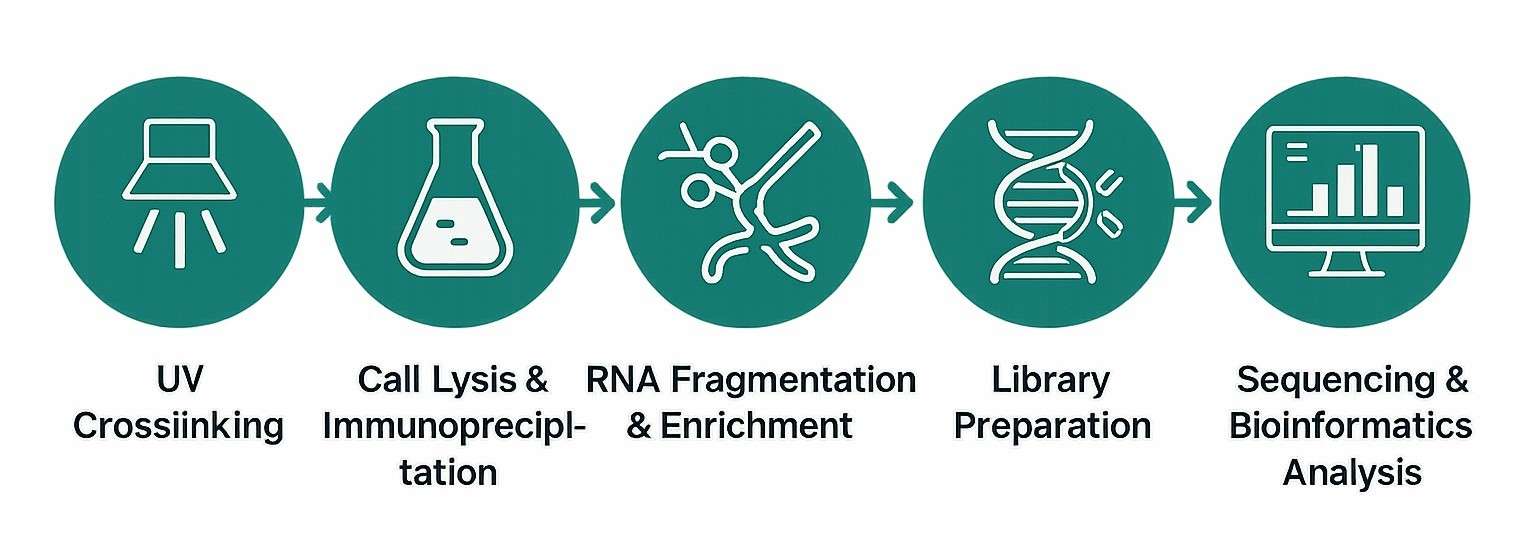
In CLIP-seq strahlen Forscher ultraviolettes (UV) Licht auf Zellen. Dadurch bleiben Proteine und RNA dort zusammen, wo sie sich berühren. Anschließend verwenden sie spezielle Antikörper, um diese Protein-RNA-Paare aus dem Rest der Zelle herauszuziehen. Danach zerlegen sie die RNA in kleine Stücke und wandeln sie in DNA um. Schließlich sequenzieren sie diese DNA, um herauszufinden, wo die Proteine an der RNA gebunden waren.
Im Vergleich zu einer einfacheren Methode, die genannt wird RIP-seq Die RNA-Immunpräzipitation-Sequenzierung (CLIP-seq) hat zwei große Vorteile:
- Höhere Spezifität: UV-Licht sorgt dafür, dass nur Proteine, die direkt mit der RNA in Kontakt stehen, verknüpft werden, was falsche Positivergebnisse reduziert.
- Bessere Auflösung: CLIP-seq zeigt genau, an welchem Teil der RNA das Protein gebunden hat, manchmal bis auf wenige Nukleotide genau.
CLIP-seq-Technologien Pakete
Klassisches CLIP-seq (HITS-CLIP)
Klassisches CLIP-seq, manchmal auch genannt HITS-CLIP, verwendet UV-Licht, um Proteine und RNA direkt in Zellen zu vernetzen. Wissenschaftler verwenden dann Antikörper, um diese Protein-RNA-Paare herauszuziehen. Anschließend wird die RNA in DNA umgewandelt und sequenziert. Diese Methode bietet eine breite, genomweite Karte wo Proteine an RNA binden.
iCLIP-seq (Einzel-Nukleotid-Auflösungs-CLIP)
iCLIP-seq ist darauf ausgelegt, Protein-RNA-Interaktionen mit Einzel-Nukleotid-PräzisionWährend des Schrittes der reversen Transkription erfasst iCLIP, wo das Enzym aufgrund von quervernetzten Proteinen ins Stocken gerät, wodurch präzise "Abschneide"-Punkte entstehen.
Diese Technologie ist besonders nützlich für das Studium von:
- Spleißregulation
- RNA-Reifung
- Funktion von nicht-kodierenden RNAs
iCLIP-seq hilft Forschern, zu identifizieren exakte Proteinbindungsstellen auf RNA, was es ideal für hochauflösende Studien macht.
miCLIP-seq (Methylierung Individuelle-Nukleotid-Auflösung CLIP)
miCLIP-seq ist eine spezialisierte Version von iCLIP, die verwendet wird, um zu studieren RNA-Modifikationeninsbesondere m6A-Methylierung.
Durch die Kombination von UV-Crosslinking mit spezifischen Antikörpern gegen methylierte Basen ermöglicht miCLIP-seq Forschern, Folgendes nachzuweisen:
- Methylierungsstellen mit Einzel-Nukleotid-Auflösung
- Bindungsstellen von "Reader"-Proteinen, die RNA-Modifikationen erkennen
Diese Technik ist entscheidend für das Verständnis. epitranskriptomische Regulation und wie chemische Veränderungen an RNA die Genexpression und Zellfunktion beeinflussen.
eCLIP-seq (Erweitertes CLIP-seq)
eCLIP-seq verbessert traditionelle CLIP-Methoden, indem es hinzufügt größenangepasste Eingabesteuerelemente um echte Bindungsereignisse besser von Hintergrundgeräuschen zu unterscheiden.
Die Vorteile von eCLIP-seq umfassen:
- Höheres Signal-Rausch-Verhältnis
- Verbesserte Reproduzierbarkeit
- Größeres Vertrauen in die Identifizierung echter Protein-RNA-Interaktionen
eCLIP-seq ist zu einer beliebten Methode für großangelegte Projekte wie ENCODE geworden, bei denen präzise und reproduzierbare Daten unerlässlich sind.
PAR-CLIP-seq
PAR-CLIP-seq besteht darin, Zellen Moleküle zuzuführen, die als photoreaktive Ribonukleosid-Analoga, wie 4-Thiouridin (4SU). Diese werden in neu transkribierte RNA eingebaut. Wenn UV-Licht die Zellen trifft, fördert es stärkere Vernetzung zwischen der RNA und den gebundenen Proteinen.
Ein einzigartiges Merkmal von PAR-CLIP ist seine Fähigkeit, genau zu bestimmen exakte Bindungsstellen auf Nukleotid-Ebene, oft erkannt durch charakteristische T-zu-C-Konversionen in den Sequenzdaten.
Vergleich von ATAC-seq, ChIP-seq, CUT&Tag, RIP-seq und CLIP-seq
Forscher fragen sich oft, welche Sequenzierungstechnologie am besten zu ihrer Studie passt. CD Genomics bietet all diese Methoden an, um Ihnen zu helfen, die Genregulation aus verschiedenen Blickwinkeln zu erkunden. Die folgende Tabelle erleichtert den Vergleich dieser leistungsstarken Werkzeuge:
| Technologie | Hauptfokus | Zielmolekül | Auflösung | Besondere Funktionen |
|---|---|---|---|---|
| ATAC-seq | Findet offene Regionen der DNA | DNA | Hoch | Schnelle Methode zur Untersuchung der Chromatinzugänglichkeit |
| ChIP-seq | Karte der Proteinbindung an DNA | DNA | Hoch | Großartig für das Studium von Transkriptionsfaktoren und Histonmodifikationen. |
| CUT&Tag | Kartiert Protein-DNA-Interaktionen mit weniger Hintergrund. | DNA | Sehr hoch | Niedrigere Eingabebedürfnisse als ChIP-seq |
| RIP-seq | Untersucht Protein-RNA-Interaktionen | RNA | Mittel | Einfacheres Protokoll, aber niedrigere Auflösung als CLIP-seq. |
| CLIP-seq | Bestimmt genaue Proteinbindungsstellen auf RNA | RNA | Sehr hoch | UV-Quervernetzung gewährleistet hohe Spezifität und Auflösung auf Nukleotid-Ebene. |
Jede Methode bietet einzigartige Einblicke. Sie können eine einzelne Technik wählen oder mehrere kombinieren, um eine tiefere Analyse zu erhalten.
"Bei CD Genomics sind wir bestrebt, durch ständige Innovation einen Schritt voraus zu sein. Wir haben tiefgehende Expertise in der Epigenomik aufgebaut, die nicht nur auf Kerntechnologien wie Methylierungsanalyse, ATAC-seq, ChIP-seq und RIP-seq fokussiert ist, sondern auch modernste Methoden wie CLIP-seq und CUT&Tag umfasst. Unser Ziel ist es, unseren Kunden zu helfen, selbst die komplexesten Herausforderungen in der epigenetischen Forschung zu bewältigen."
CLIP-seq Experimenteller Arbeitsablauf
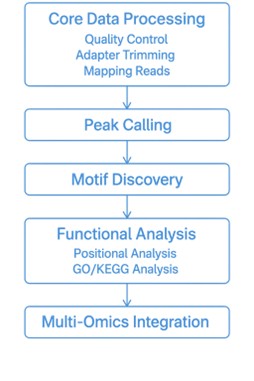
CLIP-seq Bioinformatik
Kern-Datenverarbeitung
Sobald wir die Sequenzierungsdaten erhalten, beginnen wir damit, sie aufzubereiten. Dies umfasst:
- Qualitätskontrolle: Überprüfung auf Fehler oder minderwertige Lesevorgänge, die später Probleme verursachen könnten.
- Adapter-Trimming: Entfernen von verbleibenden DNA-Stücken, die während der Bibliotheksvorbereitung verwendet wurden.
- Mapping-Lesungen: Ausrichten jedes RNA-Fragments an die richtige Stelle im Genom.
Fortgeschrittene Analyse und funktionale Interpretation
- Peak-Identifizierung: Identifizierung von Regionen, in denen viele RNA-Fragmente ansammeln, was auf eine Proteinbindestelle hinweist.
- Motiventdeckung: Suche nach kurzen RNA-Sequenzen, die Proteine bevorzugt binden.
- Positionsanalyse: Untersuchung, wo Bindungsstellen auftreten, z.B. nahe dem Anfang oder Ende von Genen (TSS, TTS, Start-/Stoppcodons).
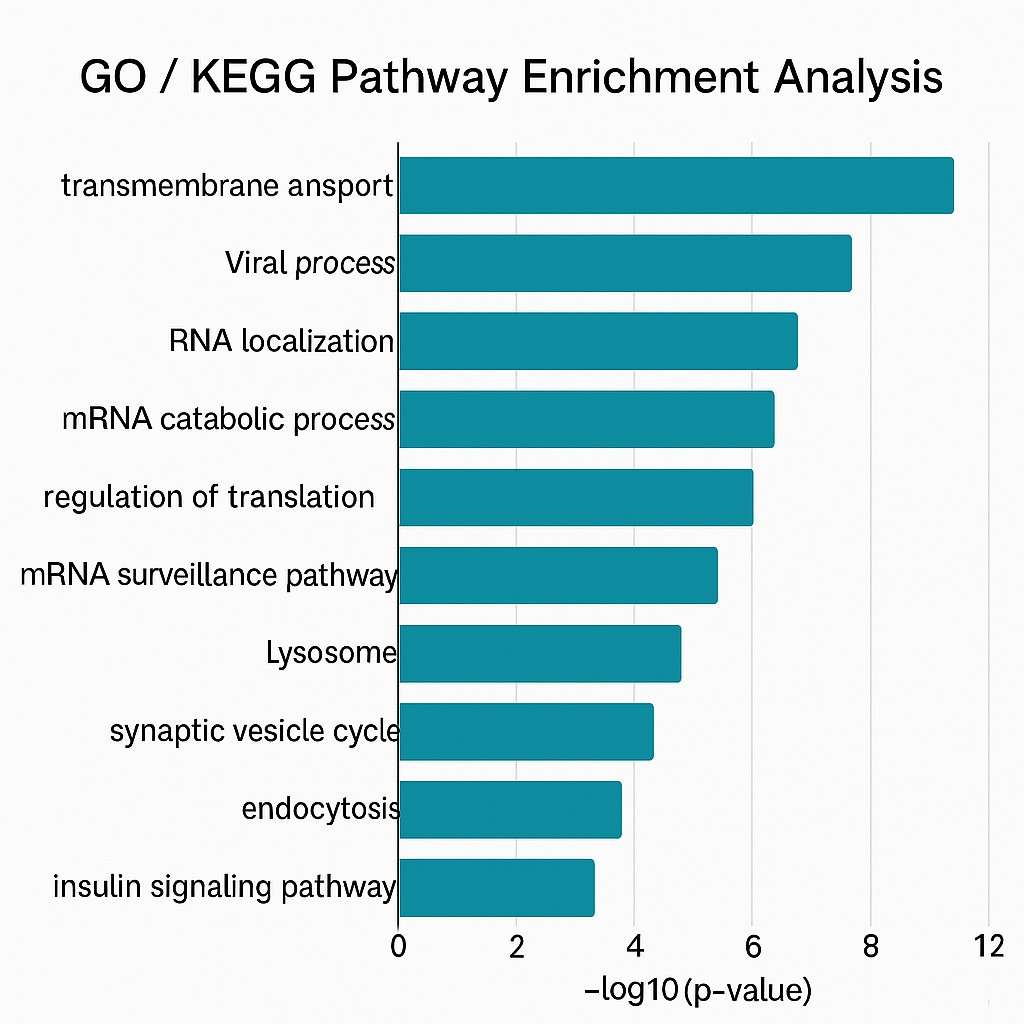
- GO- und KEGG-Pfad-Analyse: Verknüpfung gebundener Gene mit bekannten biologischen Funktionen und Wegen.
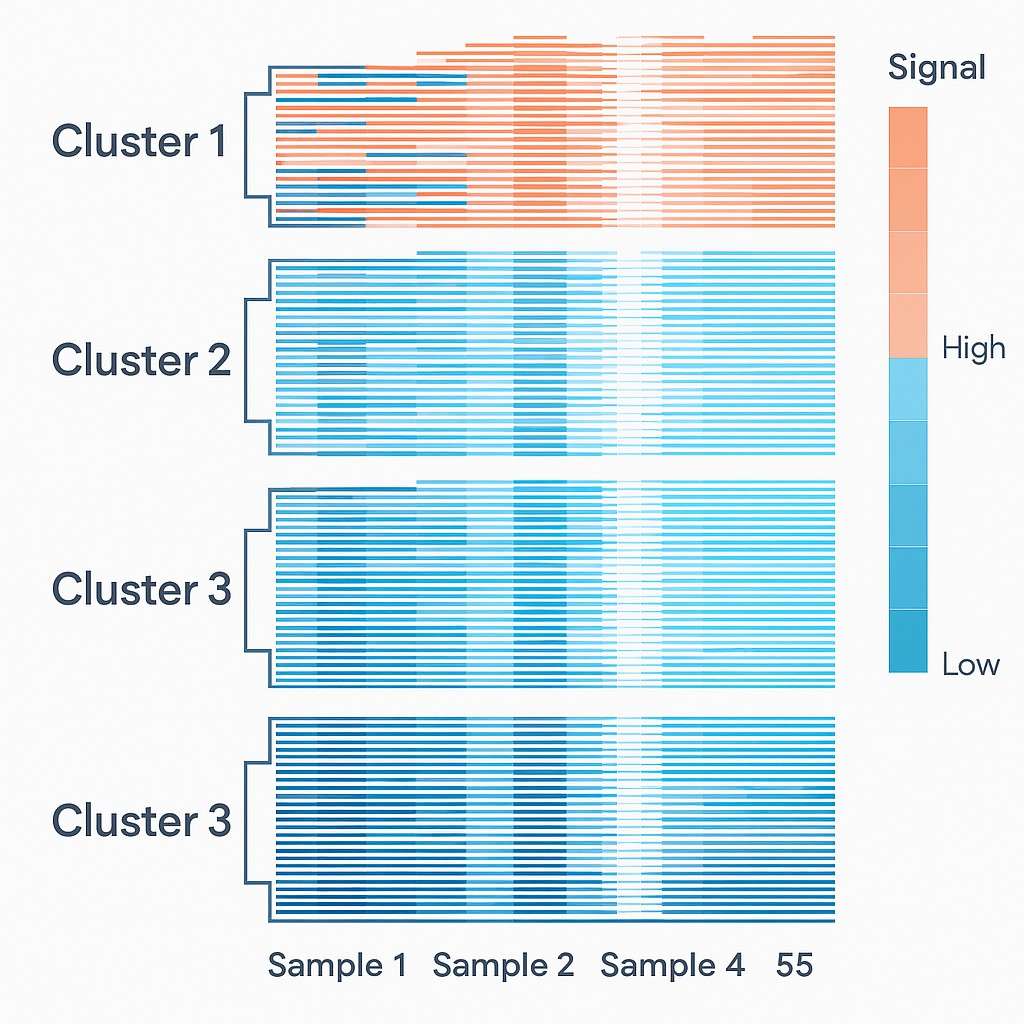
- Differentialanalyse: Vergleich von Proben, um zu sehen, wie sich die Bindung unter verschiedenen Bedingungen ändert.
- CIMS-Analyse: Erkennung winziger Veränderungen in RNA, die durch den Quervernetzungprozess verursacht werden, um Bindungsstellen genau zu bestimmen.
Wir erstellen auch klare Grafiken und Visualisierungen um Forschern zu helfen, ihre Ergebnisse zu sehen und zu interpretieren.
Multi-Omics-Integration
Moderne Forschung beinhaltet oft mehrere DatentypenCD Genomics hilft Forschern, CLIP-seq zu kombinieren mit:
- ChIP-seq: Untersuchung von Protein-DNA-Interaktionen.
- ATAC-seq: Offene Regionen der DNA finden.
- RNA-Seq: Messung der Genexpressionsniveaus.
Vorteile unserer CLIP-seq-Dienste

Umfangreiche Erfahrung mit verschiedenen Arten und Probenarten
Unser Team hat CLIP-seq-Projekte für eine Vielzahl von Organismen durchgeführt, darunter:
- Menschen
- Mäuse, Ratten und andere Tiere
- Pflanzen wie Reis, Weizen und Tomaten
- Mikroorganismen wie Bakterien und Pilze
Hohe Erfolgsquoten
CLIP-seq-Experimente können komplex sein. Aber bei CD Genomics sind unsere Die Erfolgsquoten übersteigen 95 %.dank:
- Optimierte Laborprotokolle
- Sorgfältiges experimentelles Design
- Strenge Qualitätskontrolle
Proprietäre Peak-Calling-Algorithmen
Wir haben unsere eigenen Algorithmen für entwickelt. Spitzenanruf, der entscheidende Schritt zur Identifizierung, wo Proteine an RNA binden. Unsere proprietären Methoden:
- Hintergrundgeräusche reduzieren
- Verbesserung der Erkennung von echten Bindungsereignissen
- Liefern Sie klarere, zuverlässigere Ergebnisse.
Angepasste Multi-Omics-Analysen
Jede Forschungsfrage ist anders. Deshalb bieten wir an maßgeschneiderte Datenanalyse und Multi-Omik-Integration, die es Ihnen ermöglicht:
- Verbinden Sie CLIP-seq-Daten mit ChIP-seq-, ATAC-seq- oder RNA-seq-Ergebnissen.
- Erforschen Sie komplexe regulatorische Netzwerke
- Erlangen Sie ein tieferes Verständnis der Genexpression und -regulation.
Strenge Antikörperqualitätsvalidierung
Antikörper sind die Schlüsselwerkzeuge in der CLIP-seq. Wir:
- Testen Sie jedes Antikörper auf Spezifität und Effizienz.
- Stellen Sie Validierungsdaten bereit, um Vertrauen in die Ergebnisse zu gewährleisten.
- Helfen Sie den Kunden, die richtigen Reagenzien für ihre Ziele auszuwählen.
Schnelle Bearbeitungszeiten und hochwertige Ergebnisse
Wir wissen, dass Zeit in der Forschung wichtig ist. CD Genomics bietet:
- Schnelle Projektabwicklung
- Detaillierte, veröffentlichungsbereite Berichte
- Saubere, gut organisierte Daten Dateien für eine einfache nachgelagerte Analyse
Starke Veröffentlichungshistorie
Unsere Wissenschaftler haben zu Studien beigetragen, die veröffentlicht wurden in Spitzenzeitschriften mögen Natur, Zelle, und Molekulare ZelleWir bringen dasselbe Maß an wissenschaftlicher Strenge in jedes Kundenprojekt ein.
Anwendungen von CLIP-seq
Verstehen von RNA-bindenden Proteinen (RBPs)
- Wie Gene ein- oder ausgeschaltet werden
- Wie Zellen auf Stress reagieren
- Wie verschiedene Zelltypen einzigartige Identitäten entwickeln
Untersuchung des alternativen Spleißens
- Entdecken Sie neue RNA-Varianten
- Verstehen, wie Spleißfehler zu Krankheiten beitragen
- Neue Ziele für die Arzneimittelentwicklung identifizieren
Erforschung von Nicht-kodierenden RNAs (ncRNAs)
- Entdecken Sie, wie RBPs mit nicht-kodierenden RNAs interagieren.
- Enthülle neue Funktionen für diese mysteriösen Moleküle.
- Identifizieren Sie potenzielle Biomarker für Forschungsanwendungen.
Identifizierung von miRNA-Zielen
- Hochkonfidente Identifizierung von miRNA-Zielstellen
- Einblicke, wie miRNAs spezifische biologische Wege regulieren
Unterstützung der Entdeckung von Arzneimittelzielen
- Identifizieren Sie RBPs oder RNA-Regionen, die an krankheitsbezogenen Signalwegen beteiligt sind.
- Untersuchen, wie experimentelle Medikamente RNA-Protein-Interaktionen beeinflussen.
- Finden Sie potenzielle Biomarker für die Arzneimittelentwicklung.
Beispielanforderungen
Unser HLA-Typisierungsdienst bietet hochauflösende Daten, die auf verschiedene Forschungsanwendungen zugeschnitten sind. Hier ist eine kurze Übersicht, wie man die Ergebnisse interpretiert.
| Probenart | Empfohlene Menge | Mindestmenge | Mindestkonzentration | Besondere Hinweise |
|---|---|---|---|---|
| Zellen | ≥ 1×10⁷ – 3×10⁷ Zellen | — | — | Die Anzahl hängt von der Zielproteinabundanz und der CLIP-seq-Methode ab. |
| Gewebe | ≥ 10 – 500 mg | 10 – 200 mg | — | Die Menge variiert je nach Gewebetyp und Zielprotein. Konsultieren Sie CD Genomics für Details. |
| IPed RNA | ≥ 100 ng | 40 ng | ≥ 5 ng/μL | Anwendbar, wenn gereinigte RNA nach der Immunpräzipitation eingereicht wird. |
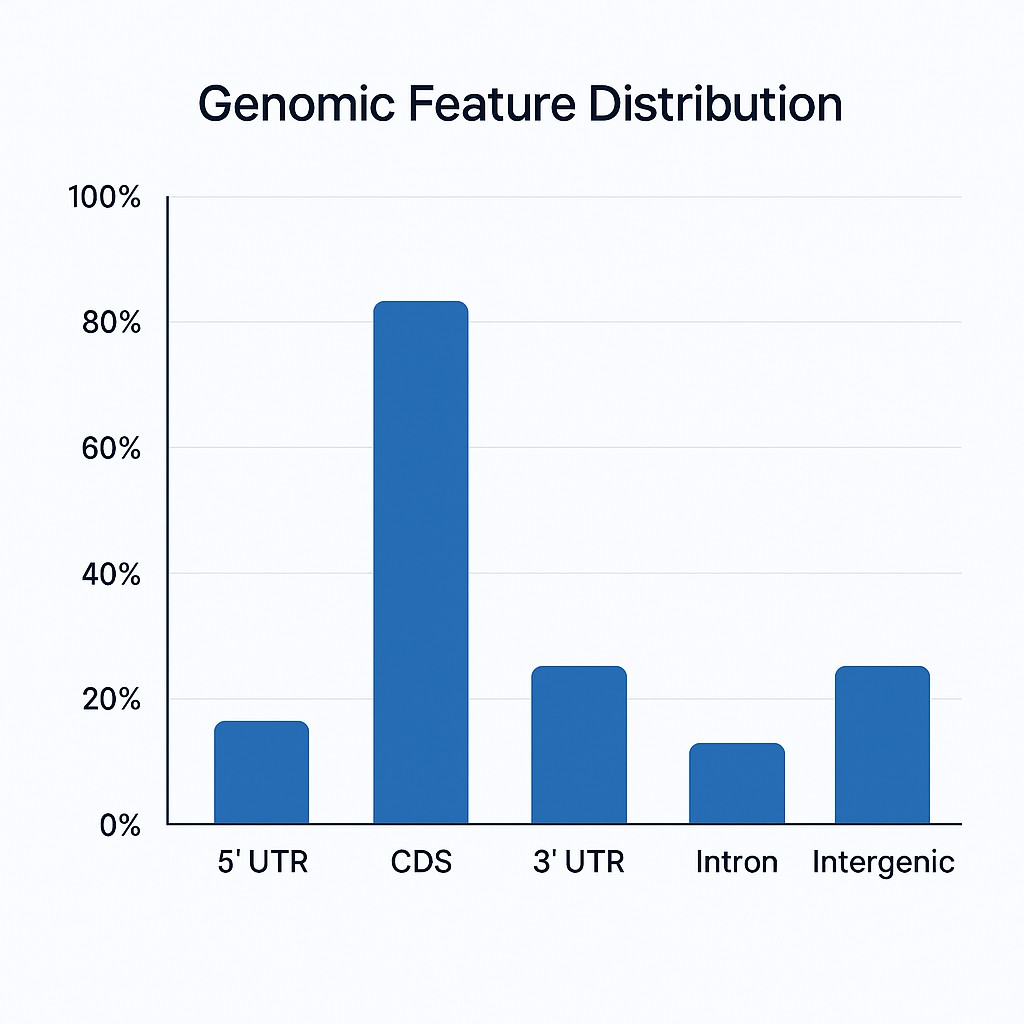
Demonstrationsergebnisse
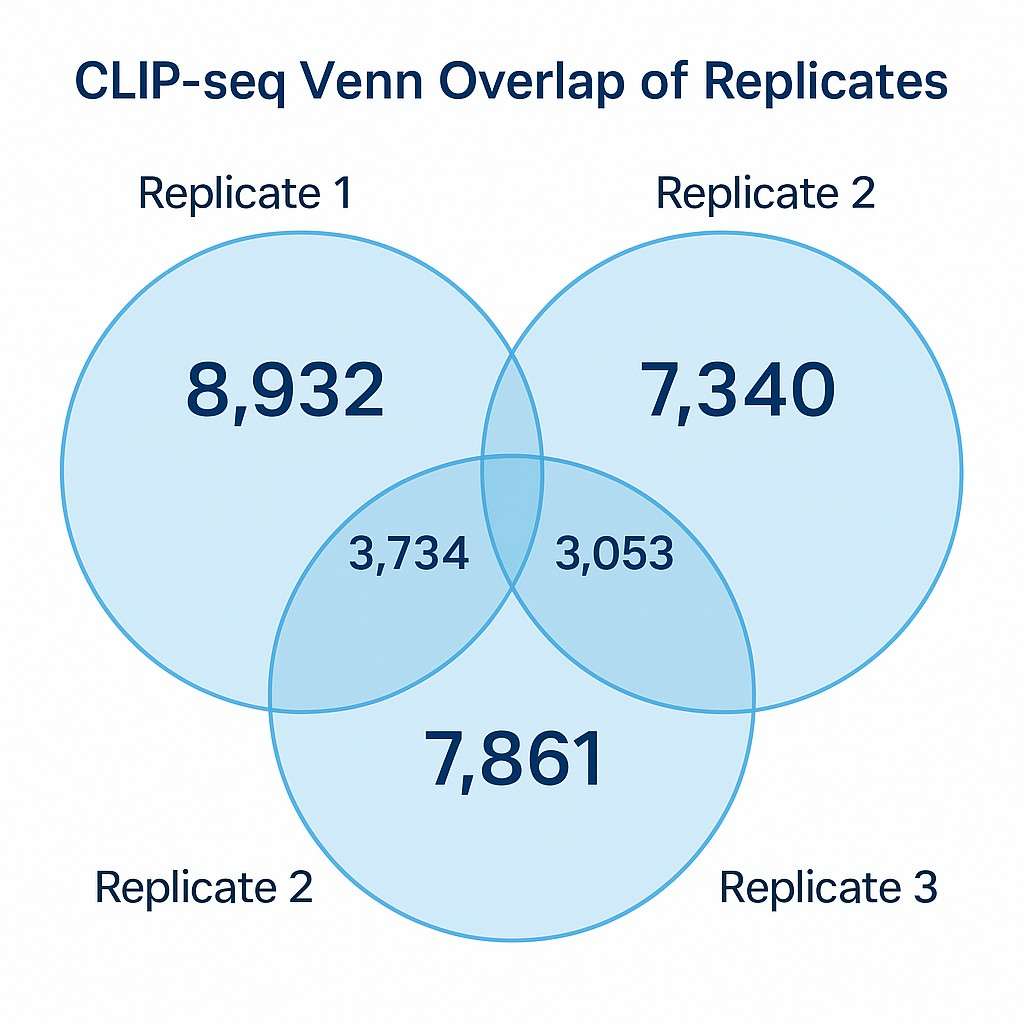
Präzise CLIP-seq FAQs
1. Was ist CLIP-seq und warum ist es wichtig?
CLIP-seq kombiniert UV-Crosslinking, Immunpräzipitation und Sequenzierung, um RNA-Protein-Interaktionen auf transkriptomweiter Ebene zu kartieren. Dies liefert präzise Einblicke, wie RNA-bindende Proteine die Genexpression posttranskriptionell regulieren.
2. Wie unterscheidet sich CLIP-seq von RIP-seq?
RIP-seq erfasst RNA-Protein-Komplexe ohne Quervernetzung, was zu mehr Hintergrundgeräuschen und weniger präziser Identifizierung von Bindungsstellen führt. CLIP-seq verwendet UV-Quervernetzung, um zu erfassen direkte, in vivo Interaktionen, anbieten höhere Spezifität und Einzel-Nukleotid-Auflösung .
3. Welche Auflösung kann ich bei CLIP-seq-Varianten wie iCLIP oder PAR-CLIP erwarten?
iCLIP erfasst truncierte cDNAs an Crosslink-Stellen, die es ermöglichen Einzel-Nukleotid-Auflösung und präzisen Protein-RNA-Bindungsort.
PAR-CLIPDie Verwendung von fotoaktivierbaren Nukleosiden führt zu charakteristischen Mutationen (z. B. T-zu-C), die dabei helfen, Bindungsstellen bis hin zu spezifischen Nukleotiden genau zu bestimmen.
4. Wie werden CLIP-seq-Daten verarbeitet und analysiert?
Typische Analysen umfassen:
- Adapterentfernung, molekulare Barcodes/Barcode-Trimmung
- Mapping von Reads auf ein Referenzgenom, Peak-Calling zur Identifizierung von Bindungsstellen.
- Motiventdeckung und Qualitätsprüfungen von Bindungsstellen
- Pipelines wie CLIPipe, PureCLIP und PEAKachu werden häufig für robuste Analyse-Workflows verwendet.
5. Welche Arten von Proben und Eingabemengen werden benötigt?
CLIP-seq erfordert typischerweise 10–30 Millionen Zellen oder 10–500 mg Gewebeje nach der spezifischen Methode. Varianten wie GoldCLIP oder irCLIP können niedrigere Eingaben akzeptieren, aber die Gesamterträge und die Proteinmenge sind entscheidende Überlegungen. Es ist am besten, CD Genomics für spezifische Ratschläge zu den Proben zu konsultieren.
Kann CLIP-seq RNA-Modifikationen wie m6A nachweisen?
Ja. Variantenmethoden wie miCLIP-seq kombinieren Sie CLIP mit Antikörpern, die auf modifizierte RNA-Basen abzielen—insbesondere m6A—um modifizierte Nukleotide mit einer Einzel-Nukleotid-Auflösung zu kartieren, was hilft, zu verstehen epitranskriptomische Regulation.
7. Wie validiere ich die Antikörperspezifität in CLIP-seq?
Hochwertiges CLIP-seq basiert auf validierten Antikörpern. Forscher sollten sicherstellen, dass ihre Antikörper spezifisch sind und gut in der Immunpräzipitation funktionieren. Werkzeuge wie Western Blot und IP-qPCR werden empfohlen, bevor mit CLIP-seq fortgefahren wird.
8. Welche Kontrollen sind in CLIP-seq-Experimenten notwendig?
Steuerungen wie Eingabemuster, größenangepasste Eingabe (SMI) für eCLIP, und IgG oder keine Antikörper-Kontrollen sind entscheidend. Diese Kontrollen helfen dabei, echte Bindungsereignisse von Hintergrundgeräuschen zu unterscheiden.
9. Wie wähle ich zwischen den CLIP-Variantenmethoden?
- Verwenden Sie iCLIP für nucleotide-genaue Kartierung.
- Wählen Sie PAR-CLIP für ein starkes Signal über photoreaktive Nukleoside.
- Betrachten Sie GoldCLIP oder irCLIP für sicherere Protokolle ohne Radioaktivität.
- Verwenden Sie eCLIP für standardisierte Eingabesteuerungen und robuste Reproduzierbarkeit.
Präzise CLIP-seq Fallstudien
Fallstudie: PRRC2B-mRNA-Interaktionen, die durch PAR-CLIP-seq kartiert wurden
Zeitschrift: Nucleic Acids Research
Impact Factor: 19,160 (2022)
Veröffentlicht: 2023
Hintergrund
RNA-bindende Proteine (RBPs) spielen eine entscheidende Rolle bei der posttranskriptionellen Regulierung der Genexpression und beeinflussen Prozesse wie Spleißen, Translation und RNA-Stabilität. Die Funktionen und Bindungsmuster vieler RBPs sind jedoch weiterhin schlecht charakterisiert. Diese Studie konzentrierte sich auf PRRC2B, ein schlecht verstandenes RBP, das verdächtigt wird, den Zellzyklus und krebsbezogene Signalwege zu beeinflussen. Die Forscher hatten das Ziel, genomweit RNA-Bindungsstellen von PRRC2B zu identifizieren und seine funktionale Rolle in der translationalen Regulation zu untersuchen.
Materialien & Methoden
Probenvorbereitung
- Zelllinie: HEK293T
- Inkorporation des photoreaktiven Ribonukleosid-Analogons 4-Thiouridin (4SU)
- UV-Quervernetzung bei 365 nm
- Immunopräzipitation & Sequenzierung
- Immunopräzipitation von PRRC2B-RNA-Komplexen
- Bibliotheksvorbereitung und PAR-CLIP-Sequenzierung
- Mapping von Reads auf das menschliche Genom
- Identifizierung von kreuzvernetzten Mutationsstellen (CIMS)
- Motivanalyse
- Ribosomen-Profilingfür Übersetzungseffizienz
- Funktionelle Assays einschließlich PRRC2B-Downregulation und Rettungsexperimenten
Ergebnisse
- Identifizierung der Bindungsstelle von PRRC2B
- Die genomweiten Bindungsstellen von PRRC2B wurden mit Einzel-Nukleotid-Auflösung kartiert.
- PRRC2B bindet bevorzugt in der Nähe der Startcodonregionen einer Untergruppe von mRNAs.
- Angereicherte Motive umfassten CU- oder GA-reiche Sequenzen in 5' UTRs und kodierenden Sequenzen.
- Funktionale Konsequenzen der PRRC2B-Bindung
- Die Knockdown von PRRC2B führte zu:
- Reduzierte Übersetzung von Ziel-mRNAs
- Verminderte Spiegel von Zellzyklus-regulatorischen Proteinen, einschließlich CCND2
- G1/S-Phase-Arrest und reduzierte Zellproliferation
- Antisense-Oligonukleotide, die die Bindung von PRRC2B an spezifische Motive stören, reduzierten die Translation von CCND2.
- Mechanistische Einblicke
- PRRC2B interagiert auf RNA-unabhängige Weise mit den Initiationsfaktoren eIF3 und eIF4G2.
- Mutationen, die die Interaktionen von PRRC2B mit Initiationsfaktoren stören, konnten die durch PRRC2B-Downregulation verursachten Übersetzungsdefekte nicht beheben.
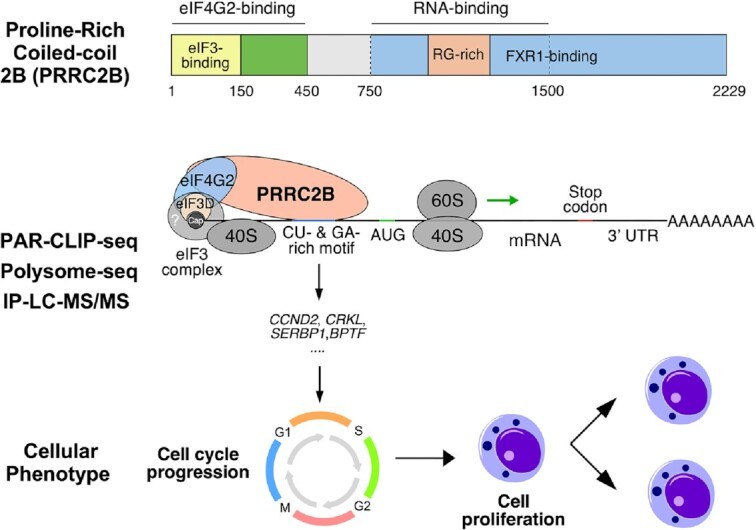 Prolinreiches Coiled-Coil-Protein 2B (PRRC2B) bindet an CU- oder GA-reiche Motive auf spezifischen mRNAs, erleichtert deren Translation durch Interaktion mit eIF4G2 und eIF3 und fördert den Zellzyklus und die Zellproliferation.
Prolinreiches Coiled-Coil-Protein 2B (PRRC2B) bindet an CU- oder GA-reiche Motive auf spezifischen mRNAs, erleichtert deren Translation durch Interaktion mit eIF4G2 und eIF3 und fördert den Zellzyklus und die Zellproliferation.
Fazit
Diese Studie bietet die erste umfassende, hochauflösende Karte der PRRC2B-RNA-Interaktionen in menschlichen Zellen und zeigt, dass PRRC2B in der Nähe von Translationstartstellen bindet und die Translation wichtiger Zellzyklusregulatoren verbessert. Die Störung dieser Interaktionen beeinträchtigt den Zellzyklusfortschritt und deutet auf einen neuartigen Mechanismus der translationalen Kontrolle hin. Diese Erkenntnisse heben PRRC2B als potenziellen regulatorischen Knotenpunkt in der Krebsbiologie und den Zellproliferationswegen hervor.
Referenz
- Jiang, F., Hedaya, O. M., Khor, E. S., et al. (2023). Das RNA-bindende Protein PRRC2B vermittelt die Translation spezifischer mRNAs und reguliert den Zellzyklus. Nukleinsäurenforschung. PMC10287950


