
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
DAP-Seq-Dienst (DNA-Affinitätsreinigung-Sequenzierung)
CD Genomics bietet DAP-seq-Dienste an, die moderne genomische Technologien nutzen, um die Bindung von Transkriptionsfaktoren zu analysieren. Unser Hochdurchsatzansatz identifiziert TF-DNA-Interaktionen und liefert Einblicke in genregulatorische Netzwerke in verschiedenen Forschungsbereichen. Dieser Service eignet sich hervorragend, um die Mechanismen der Genexpression sowohl in Pflanzen als auch in Tieren zu entschlüsseln und Fortschritte in der biologischen Forschung zu ermöglichen.
Die Einführung von DAP-Seq
Die umfassende Identifizierung von TFBS (Transkriptionsfaktor-Bindungsstellen) im gesamten Genom ist entscheidend für die Charakterisierung regulatorischer Elemente und der Funktion von Transkriptionsfaktoren. ChIP-Seq (Chromatin-Immunpräzipitation-Sequenzierung) ist ein leistungsstarker Ansatz zur Bestimmung von in vivo TFBS. Die Methode ist jedoch in ihrem Umfang begrenzt, da sie auf spezifischen Antikörpern für jeden Transkriptionsfaktor basiert, was technisch herausfordernd und kostspielig sein kann. Derzeit können alternative Methoden wie DAP-Seq eine einfache und hochdurchsatzfähige Methode bieten, um die Bindung von TF an sein entsprechendes Ziel (gDNA) in einem chromatinfreien Kontext zu untersuchen, während wichtige Informationen zu primären Genomsequenzen und DNA-Methylierung erhalten bleiben. Die Methode erfordert keine spezifischen Antikörper oder gen-spezifischen Primer, ist für alle Eukaryoten geeignet und stellt ein leistungsstarkes Werkzeug zum Verständnis von regulatorischen Elementen und der Funktion von TF dar.
Der vollständige Name von DAP-seq steht für DNA-Affinitätsreinigung-Sequenzierung. Diese Technik beinhaltet die Identifizierung von Transkriptionsfaktor-Bindungsstellen (TFBS) durch die in vitro Expression von Transkriptionsfaktoren, ohne durch Antikörper oder Spezies eingeschränkt zu sein, und bietet Vorteile in der Hochdurchsatztechnologie. Seit ihrer Einführung wurde diese Technologie umfassend in der Untersuchung der transkriptionalen Regulation angewendet und Epigenomik.
Prinzip des DAP-seq-Experiments:
In DAP-seq-Experimenten werden das extern exprimierte Protein und die DNA einer Affinitätsreinigung unterzogen, gefolgt von Hochdurchsatz-Sequenzierung der DNA, die an das Protein gebunden ist. Der grundlegende Prozess umfasst den Aufbau der kodierenden Sequenz (CDS) des Transkriptionsfaktors in einen Vektor, der ein Affinitäts-Tag enthält, die Erstellung eines Protein-Expressionsvektors und die Durchführung einer in vitro-Proteinexpression, um ein Fusionsprotein des Transkriptionsfaktors und des Affinitäts-Tags zu produzieren. Anschließend wird genomische DNA aus der Probe extrahiert, eine DNA-Bibliothek erstellt und der in vitro exprimierte Transkriptionsfaktor mit dem Affinitäts-Tag wird mit der DNA-Bibliothek kombiniert. Die gebundene DNA wird dann eluiert und einer Sequenzierung unterzogen.
Vergleich von DAP-seq und ChIP-seq Technologien
| Technologie Name | DAP-seq | ChIP-seq |
|---|---|---|
| Experimenteller Modus | In vitro | In vivo |
| Erfordert spezifische Antikörper | Nein | Ja |
| Geeignet für Nicht-Modellorganismen | Ja | Nein |
| Zeitkosten | Niedrig | Hoch |
| Hohe Durchsatzleistung | Ja | Nein |
Vorteile von DAP-Seq
- Geeignet für die Hochdurchsatz-Probenverarbeitung ganzer TF ORF Klonkollektionen.
- Erfordert nicht die spezifischen Antikörper für jeden Transkriptionsfaktor, ist relativ kostengünstig und für alle Eukaryoten geeignet.
- DAP-Seq behält viele der gewebespezifischen/zelllinien-spezifischen sekundären Modifikationen und Merkmale bei, die im gesamten Genom vorhanden sind.
- Der Anreicherungsprozess von DNA-Fragmenten an TF-Bindungsstellen ist einfacher.
Anwendungen von DAP-Seq
- Analyse und funktionale Charakterisierung von Transkriptionsfaktor-Bindungsstellen
- Identifizierung von Histonmodifikationstypen an spezifischen DNA-Loci
- Studie über die Korrelation zwischen Histonmodifikationen und Genexpression
DAP-Seq-Workflow
Unser hochqualifiziertes Expertenteam führt das Qualitätsmanagement nach jedem Verfahren durch, um umfassende und genaue Ergebnisse zu gewährleisten. Unser DAP-Seq-Workflow ist unten aufgeführt, einschließlich Bibliotheksvorbereitung, Proteinexpression, DNA-Affinitätsreinigung, Sequenzierung und bioinformatischer Analyse.
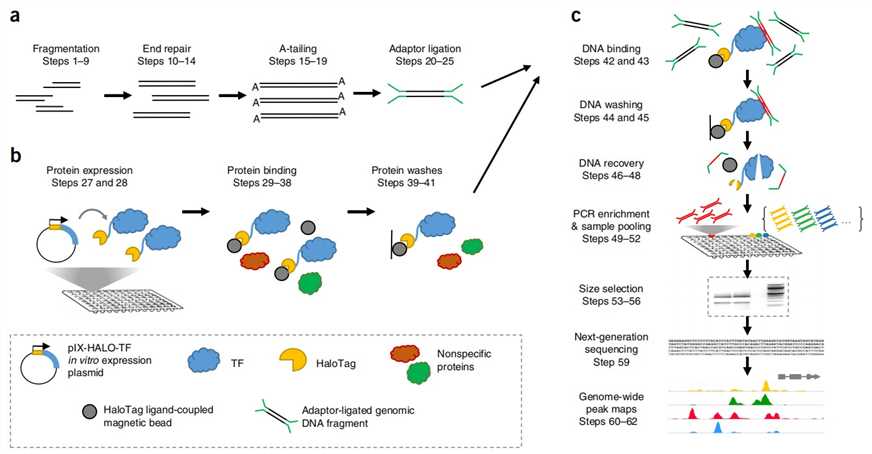 Abb. 1. Experimentelles Verfahren von DAP-seq. (Anna Bartlett et al., 2017)
Abb. 1. Experimentelles Verfahren von DAP-seq. (Anna Bartlett et al., 2017)
Dienstspezifikationen
Beispielanforderungen
|
|
 Klicken |
Sequenzierungsstrategie
|
 |
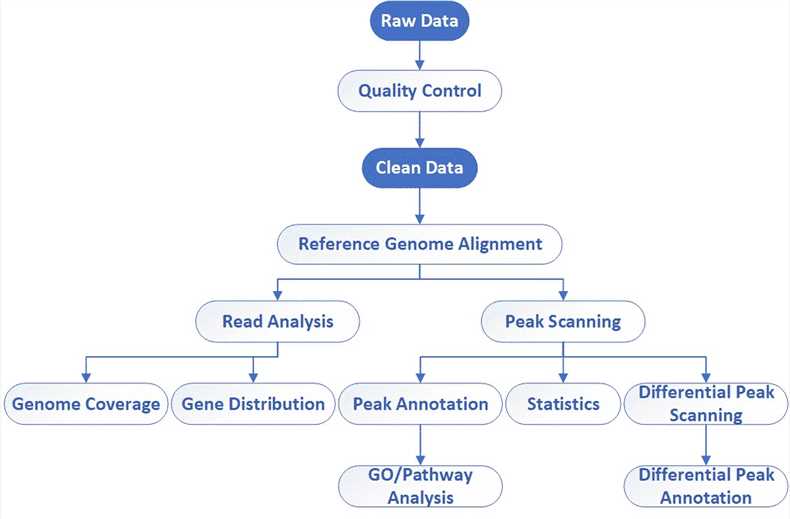
Bioinformatische Analyse Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an:
|
Analyse-Pipeline
Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details in DAP-Seq für Ihr Schreiben (Anpassung)
Bei CD Genomics bieten wir Ihnen hochwertige Sequenzierung und umfassende bioinformatische Analysen für Ihr DAP-Seq-Projekt an, die die Identifizierung von TFBS (Transkriptionsfaktor-Bindungsstellen) im gesamten Genom ermöglichen. Wenn Sie zusätzliche Anforderungen oder Fragen haben, zögern Sie bitte nicht, uns zu kontaktieren.
Referenzen
- Ronan C. O'Malley, S. Shan C. Huang, L. Song u. a. Cistrome- und Epicistrome-Merkmale gestalten die regulatorische DNA-Landschaft, Zelle, 165 (2016) 1280–1292.
- Anna Bartlett, Ronan C. O'Malley, S. shan C. Huang, et al. Kartierung von genombreiten Bindungsstellen für Transkriptionsfaktoren mittels DAP-Seq, Naturprotokolle, 12 (2017) 1659–1672.
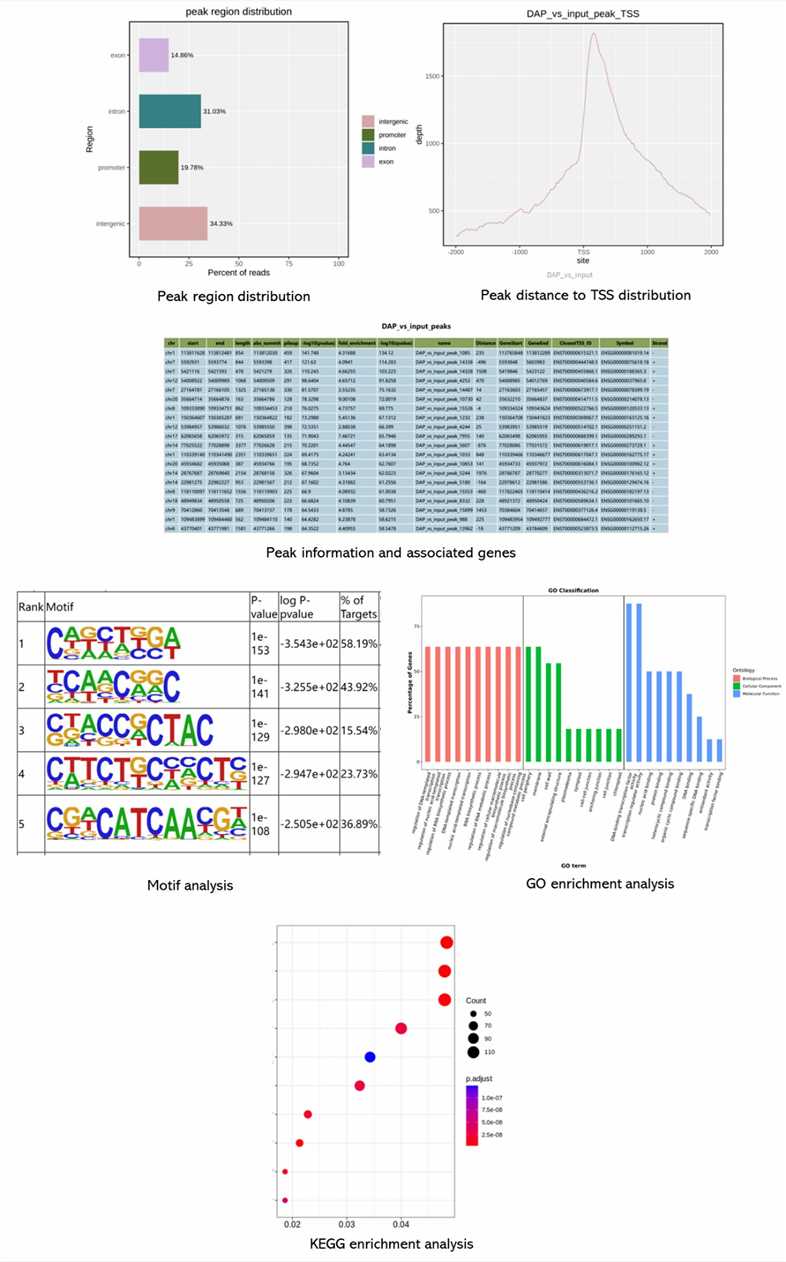
Demonstrationsergebnisse
Teilweise Ergebnisse sind unten aufgeführt:
DAP-Seq (DNA-Affinitätsreinigung Sequenzierung) FAQs
1. Wie vereinfacht DAP-seq die Anreicherung von TF-Bindungsstellen?
DAP-seq vereinfacht die Anreicherung, indem Transkriptionsfaktoren (TFs) mit einem Halo-Tag versehen und magnetische Perlen verwendet werden, die mit liganden spezifisch für den Tag beschichtet sind. Diese magnetische Pull-Down-Strategie ersetzt die traditionelle Immunpräzipitation und reduziert die experimentelle Komplexität.
Kann DAP-seq DNA-Bindungsstellen von regulatorischen Faktoren nachweisen?
Einige regulatorische Faktoren binden indirekt an DNA über Transkriptionsfaktoren, und DAP-seq kann nur Bindungsstellen von Transkriptionsfaktoren erkennen, die direkt an DNA binden. Daher wird nicht empfohlen, DAP-seq zu verwenden, um Bindungsstellen von regulatorischen Faktoren zu erkennen, die indirekt an DNA binden.
3. Gibt es Einschränkungen bei DAP-seq?
Trotz seiner Vorteile basiert DAP-seq auf der in vitro-Proteinexpression, was einige Unterschiede im Vergleich zu vollständig in vivo-Experimenten mit sich bringen kann.
4. Wie unterscheidet sich DAP-seq von In-vivo-Experimenten?
In vitro-Bedingungen unterstützen möglicherweise nicht die ordnungsgemäße Protein-Faltung oder die komplexe Bildung, die für die TF-DNA-Bindung erforderlich sind, was die Funktionalität einiger TFs beeinträchtigen kann.
DAP-Seq (DNA-Affinitätsreinigung Sequenzierung) Fallstudien
Ein Brassinosteroid-Transkriptionsregulationsnetzwerk ist an der Regulierung der Faserelastizität in Baumwolle beteiligt.
Zeitschrift: Pflanzenphysiologie
Impact-Faktor: 7,479
Veröffentlicht: 21. Dezember 2022
Hintergrund
Brassinosteroide (BRs) sind essentielle Substanzen, die in Raps vorkommen und verschiedene Entwicklungs- und physiologische Prozesse im Pflanzenreich regulieren. Ein Mangel oder eine Unempfindlichkeit gegenüber BRs führt zu erheblichen morphologischen Veränderungen. Bei Baumwolle spielen BRs eine entscheidende Rolle bei der Faserelastizität, indem sie die Genexpression im Zusammenhang mit der Biosynthese der Zellwand und der Dynamik des Zytoskeletts beeinflussen. Der Transkriptionsfaktor GhBES1.4 reguliert positiv die Elongation der Baumwollfaser, indem er an spezifische Zielgene bindet, wie durch DAP-seq identifiziert wurde und RNA-Seq Analysen.
Materialien & Methoden
Probenvorbereitung
- Baumwolle
- Gewebeproben
- gDNA-Extraktion
- RNA-Extraktion
Sequenzierung
- Bibliotheksbau
- RNA-Seq
- DAP-seq
- Transkriptionale Analyse von differentiellen Genen
- Identifizierung von Zielgenen
- GWAS Analysen
- Statistische Analyse
Ergebnisse
In dieser Studie verwendeten die Autoren DAP-seq, um 9.894 hochkonfidente Bindungsregionen von GhBES1.4 in Baumwolle zu identifizieren, die überwiegend in der Nähe von Transkriptionsstartstellen lokalisiert sind. Sie charakterisierten weiter die Bindungsmotive von GhBES1.4 und zeigten eine Präferenz für E-Box-Elemente sowie eine Bindung an BRRE und neuartige Motive (MEME-3/4/5). Hefe-Ein-Hybrid- und BLI-Assays bestätigten die Affinität von GhBES1.4 zu diesen Motiven und hoben seine Rolle bei der Regulierung der Genexpression hervor, einschließlich GhCPD, GhCYP90D1 und GhBRL, die entscheidend für die Entwicklung von Baumwollfasern sind.
 Abbildung 1 Identifizierung der direkten Zielgene von GhBES1.4 mittels DAP-seq.
Abbildung 1 Identifizierung der direkten Zielgene von GhBES1.4 mittels DAP-seq.
 Abbildung 2 Charakterisierung der Zielstelle für GhBES1.4.
Abbildung 2 Charakterisierung der Zielstelle für GhBES1.4.
Die Autoren erzeugten GhBES1.4-OE- und RNAi-transgene Baumwollpflanzen und fanden heraus, dass GhBES1.4 die Faserelastizität positiv reguliert. GhBES1.4-OE-Pflanzen zeigten eine erhöhte Faserlänge und eine Herabregulierung von BR-verwandten Genen, während RNAi-Pflanzen das Gegenteil aufwiesen. Die RNA-Seq-Analyse ergab 1.788 differentielle exprimierte Gene (DEGs) in GhBES1.4-OE und 1.566 in RNAi-Pflanzen, die Gene betreffen, die mit der Zellwandeelongation, der Synthese der Primärwand und der Tubulinassemblierung in Verbindung stehen, was auf die Rolle von GhBES1.4 bei der Regulierung der Genexpression, die mit der Faserelastizität in Zusammenhang steht, hinweist.
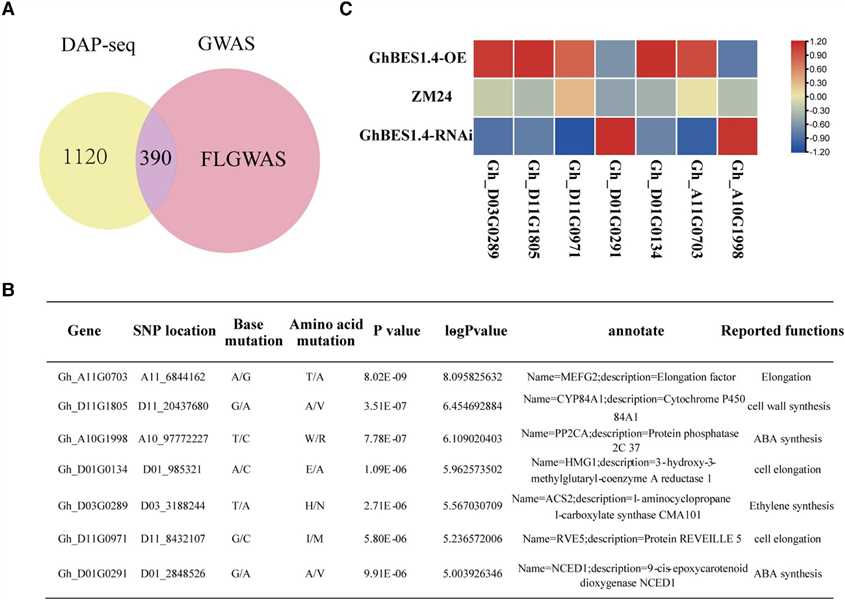 Abbildung 3 Die Integration von DAP-seq, RNA-seq und genomweiten Assoziationsstudien (GWAS) identifiziert die Zielgene für GhBES1.4 zur Regulierung der Faserelastizität.
Abbildung 3 Die Integration von DAP-seq, RNA-seq und genomweiten Assoziationsstudien (GWAS) identifiziert die Zielgene für GhBES1.4 zur Regulierung der Faserelastizität.
Diese Studie integrierte DAP-seq, RNA-seq und GWAS, um die Zielgene von GhBES1.4 zu identifizieren, die die Verlängerung von Baumwollfasern regulieren. GWAS identifizierte 390 Gene mit SNP-Loci, von denen 35 stark mit der Faserlänge (FL) assoziiert waren. Die Transkriptomanalyse und qPCR bestätigten die unterschiedliche Expression von 7 Genen in transgenen Materialien. Biofilm-Interferenzassays bestätigten die Bindung von GhBES1.4 an die Promotoren der Kandidatengene, und LUC-Aktivitätsassays zeigten die direkte Aktivierung durch GhBES1.4. Diese Gene, einschließlich RVE5, HMG1 und CYP84A1, beeinflussen die Faserentwicklung über Wege wie Zellverlängerung und Zellwandsynthese. Das Herunterregulieren von GhCYP84A1 und GhHMG1 verkürzte die Faserlänge signifikant und unterstrich deren Bedeutung in der von GhBES1.4 vermittelten Regulation der Verlängerung von Baumwollfasern.
 Abbildung 4 Mechanismus der von GhBES1.4 vermittelten Faserelastizität.
Abbildung 4 Mechanismus der von GhBES1.4 vermittelten Faserelastizität.
Fazit
Diese Studie zeigt die Wirksamkeit von DAP-seq zur Identifizierung von GhBES1.4-Zielgenen in Baumwolle und enthüllt 1.531 potenzielle Ziele. Dieser Ansatz bietet eine schnelle und robuste Methode, um TF-vermittelte regulatorische Netzwerke in Pflanzen wie Baumwolle zu entschlüsseln.
Referenz
- Liu L, Chen G, Li S, et al. Ein Transkriptionsregulationsnetzwerk von Brassinosteroiden ist an der Regulierung der Faserelastizität in Baumwolle beteiligt. Pflanzenphysiologie2023, 191(3):1985-2000.
Verwandte Veröffentlichungen
Hier sind einige Veröffentlichungen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Die Spaltung von Phagen-DNA durch Restriktionsendonukleasen ermöglicht die Wiederbelebung aus der durch Cas13 induzierten bakteriellen Dormanz.
Zeitschrift: Nature Mikrobiologie
Jahr: 2023
IL-4 fördert die Erschöpfung von CD8.+ CART-Zellen
Zeitschrift: Nature Communications
Jahr: 2024
Fettreiche Diäten während der Schwangerschaft führen zu Veränderungen der DNA-Methylierung und Proteinexpression im Pankreasgewebe des Nachwuchses: Ein Multi-Omics-Ansatz
Zeitschrift: Internationale Zeitschrift für Molekulare Wissenschaften
Jahr: 2024
KMT2A assoziiert mit dem PHF5A-PHF14-HMG20A-RAI1 Subkomplex in Stammzellen des Pankreaskarzinoms und reguliert epigenetisch deren Eigenschaften.
Zeitschrift: Nature Communications
Jahr: 2023
Krebsassoziierte DNA-Hypermethylierung von Polycomb-Zielen erfordert die doppelte Erkennung von Histon H2AK119-Ubiquitinierung und der sauren Tasche des Nukleosoms durch DNMT3A.
Journal: Wissenschaftliche Fortschritte
Jahr: 2024
Genomisches Imprinting-ähnliches monoalleles väterliches Ausdrucksmuster bestimmt das Geschlecht von Kanalkatzenfischen.
Journal: Wissenschaftliche Fortschritte
Jahr: 2022
Mehr ansehen Artikel, die von unseren Kunden veröffentlicht wurden.


