
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
2'-O-RNA-Methylierungs-Sequenzierungsdienst
CD Genomics bietet hochmoderne 2'-O-RNA-Methylierungssequenzierungsdienste an, die speziell entwickelt wurden, um 2'-O-Methylierungsmuster in RNA-Molekülen zu erkennen und zu analysieren. Unsere Methodik bietet hohe Sensitivität und Genauigkeit, die eine präzise Identifizierung von methylieren Nukleotiden ermöglicht. Dieser Dienst unterstützt umfassende Studien zur RNA-Struktur, Stabilität und regulatorischen Rollen in biologischen Prozessen.
Was ist die 2'-O-Methylierung von RNA?
2'-O-Methylierung (Nm) ist eine posttranskriptionale Modifikation von RNA, die durch 2'-O-Methyltransferasen katalysiert wird und den Austausch eines Wasserstoffatoms an der 2-Hydroxylgruppe gegen eine Methylgruppe beinhaltet. Diese chemische Modifikation tritt an der 2'-Position von Ribose in RNA auf und wird von RNA-Methyltransferasen oder Fibrillarin vermittelt. Bemerkenswerterweise ist die 2'-O-RNA-Methylierung eine Modifikation, die ubiquitär in verschiedenen Arten vorkommt, einschließlich Säugetieren, Hefen, Pflanzen und Viren.
Jüngste Studien haben gezeigt, dass 2'-O-RNA-Methylierung auf verschiedenen RNA-Molekülen wie mRNA, tRNA, rRNA und miRNA vorhanden ist. Strukturell erhöht diese Modifikation die Hydrophobizität des Nukleotids und steigert somit dessen Stabilität. Die funktionalen Implikationen der 2'-O-RNA-Methylierung wurden in mehreren biologischen Prozessen aufgeklärt; sie beeinflusst die mRNA-Protein-Interaktionen, reguliert die Übersetzungseffizienz von rRNA und beteiligt sich an der Erkennung von tRNA.
Transfer-RNA (tRNA) weist eine umfangreiche Reihe von posttranskriptionalen Modifikationen auf, die entscheidend für die Ausübung ihrer biologischen Funktionen sind. Unter diesen sticht die Methylierung als die häufigste Art der tRNA-Modifikation hervor, die entweder an den Nukleotidbasen oder an der 2'-O-Ribose-Position (2'-O-Methylierung) erfolgt. Die 2'-O-Methylierungsmodifikation ist in Archaeen, Prokaryoten und Eukaryoten weit verbreitet und wird an den Positionen 4, 6, 18, 32, 34, 39, 44, 54 und 56 innerhalb der tRNA identifiziert.
Forschungsergebnisse deuten darauf hin, dass die 2'-O-Methylierung eine regulatorische Rolle bei der Faltung, Reifung, Stabilität und der Präzision der Anticodon-mRNA-Codon-Paarung von tRNA spielt. Sie ist eng mit zellulären Prozessen wie Wachstum, Stressreaktion und Immunregulation verbunden. Mängel an zytoplasmatischer tRNA-2'-O-Methylierung werden häufig mit verschiedenen Krankheiten in Verbindung gebracht. Enzyme, die an der tRNA-2'-O-Methylierung beteiligt sind, stellen potenzielle Ziele für die Arzneimittelentwicklung dar.
Die Untersuchung der 2'-O-Methylierung von tRNA und ihrer modifizierenden Enzyme wird unser Verständnis der biologischen Funktionen dieser Modifikation erweitern. Darüber hinaus legt sie die Grundlage für die Erforschung der pathogenetischen Mechanismen der tRNA-2'-O-Methylierungsenzyme bei menschlichen Krankheiten.
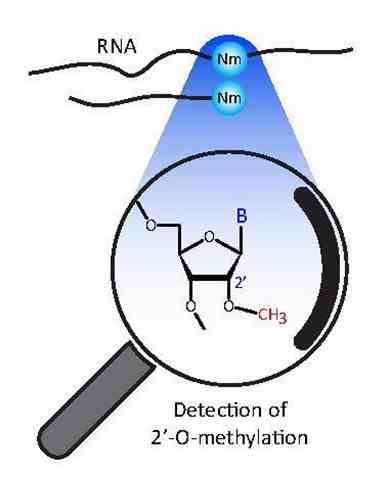 Abbildung 1. Nachweis von RNA-Ribose-2'-O-Methylierungen (Yuri Motorin, Virginie Marchand, 2018)
Abbildung 1. Nachweis von RNA-Ribose-2'-O-Methylierungen (Yuri Motorin, Virginie Marchand, 2018)
Die Einführung der 2'-O-RNA-Methylierungs-Sequenzierung
Um weitere 2'-Methylierungsmodifikationen zu erkennen und weitere biologische Funktionsmechanismen zu klären, wurden zahlreiche biologische experimentelle Techniken vorgeschlagen. Dazu gehören RNase H-basierte Methoden, reverse Transkriptionsansätze und PCR-basierte Strategien. Diese Methoden sind jedoch oft zeitaufwendig. Mit dem Fortschritt der Sequenzierungstechnologien nimmt die Ansammlung von Nukleotidsequenzen weiter zu. Der Kern der 2'-O-RNA-Methylierungssequenzierung besteht darin, die 2'-O-Methylierungsstellen innerhalb von RNA-Molekülen mithilfe chemischer oder enzymatischer Methoden zu identifizieren, gefolgt von einer präzisen Lokalisierung und Quantifizierung dieser Stellen durch Hochdurchsatz-Sequenzierung Techniken. Die 2'-O-Methylierung verändert die chemischen Eigenschaften von RNA-Molekülen und ermöglicht die Identifizierung und Lokalisierung dieser Modifikationen.
Derzeit werden mehrere Methoden hauptsächlich für die 2'-O-RNA-Methylierungs-Sequenzierung eingesetzt:
- Ribose-seqDiese Technik konzentriert sich auf ribosomale RNA (rRNA). Sie umfasst den gezielten enzymatischen Schnitt von RNA-Ketten, um 2'-O-methylierte Ribose freizusetzen, die anschließend weiterverarbeitet wird. Hochdurchsatz-Sequenzierung.
- Nm-seqDieses Verfahren verwendet spezifische chemische Modifikationen, um 2'-O-Methylierungsstellen zu kennzeichnen. Diese Modifikationen ermöglichen die Erkennung von Methylierungsstellen durch reversen Transkriptionsreaktionen, gefolgt von Hochdurchsatz-Sequenzierung.
- Pseudouridin-Sequenzierung (Ψ-seq)Obwohl diese Methode hauptsächlich zur Identifizierung von Pseudouridin (Ψ)-Modifikationen entwickelt wurde, kann sie mit anderen Nachweistechniken kombiniert werden, um das Studium der 2'-O-Methylierung zu erleichtern.
CD Genomics bietet einen umfassenden Service zur Erkennung von 2'-O-RNA-Methylierung an, der darauf ausgelegt ist, das Vorhandensein von 2'-O-Methylierungsmodifikationen an RNA-Molekülen zu identifizieren und die spezifischen Stellen der 2'-O-Methylierung zu bestimmen. Neben dem angebotenen Service hat CD Genomics eine Reihe von Produkten entwickelt, die zur Bewertung der 2'-O-RNA-Methylierungsniveaus in verschiedenen RNA-Molekülen dienen, einschließlich mRNA, LncRNA, pri-miRNA, tRNA und rRNA.
Ansätze zur tiefen Sequenzierung zur Erkennung von 2'-O-Methylierung
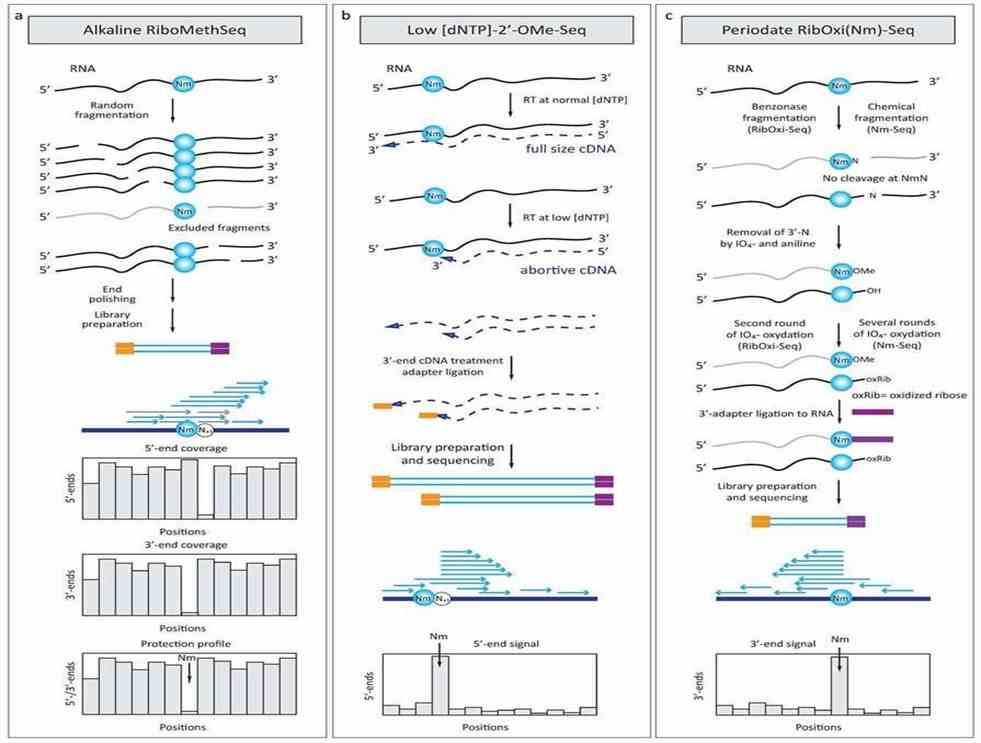 Abbildung 2. Auf tiefen Sequenzierung basierende Ansätze zur Detektion von 2'-O-methylierte Rückstände in RNA (Yuri Motorin, Virginie Marchand, 2018)
Abbildung 2. Auf tiefen Sequenzierung basierende Ansätze zur Detektion von 2'-O-methylierte Rückstände in RNA (Yuri Motorin, Virginie Marchand, 2018)
Vorteile unseres 2'-O-RNA-Methylierungs-Sequenzierungsdienstes
- Individuelle NukleotidgenauigkeitUnser Service zeichnet sich durch die präzise Identifizierung von 2'-O-RNA-Methylierungsmodifikationen mit Einzel-Nukleotid-Präzision aus. Dieser anspruchsvolle Ansatz gewährleistet eine sorgfältige Untersuchung der Modifikationsstandorte innerhalb von RNA-Molekülen.
- Effiziente HochdurchsatzdetektionDurch den Einsatz fortschrittlicher Hochdurchsatztechniken gewährleisten wir die parallele und effiziente Identifizierung von 2'-O-RNA-Modifikationsstellen im gesamten Transkriptom. Diese Strategie ermöglicht die gleichzeitige Bewertung mehrerer RNA-Moleküle und liefert einen umfassenden Überblick über die 2'-O-RNA-Methylierungsmuster.
- Umfassende Abdeckung über RNA-Arten hinwegUnser Service umfasst eine Vielzahl von RNA-Typen, darunter mRNA, LncRNA, pri-miRNA, tRNA und rRNA. Diese umfassende Analyse gewährleistet die Identifizierung von 2'-O-RNA-Methylierungsstellen in verschiedenen RNA-Molekülen und trägt zu einem nuancierten Verständnis der Modifikationslandschaft bei.
- Spezialisierte bioinformatische InterpretationUnterstützt von einem kompetenten Bioinformatik-Team bieten wir spezialisierte Datenanalyse-Dienstleistungen, die auf die unterschiedlichen Anforderungen unserer Kunden zugeschnitten sind. Unser Team ist erfahren in der Durchführung umfassender Datenanalysen und bietet aufschlussreiche Interpretationen der Ergebnisse, die aus 2'-O-RNA-Methylierungsnachweisergebnissen gewonnen werden.
Anwendungen der 2'-O-RNA-Methylierung-Sequenzierung
2'-O-RNA-Methylierung-Sequenzierung hat wichtige Anwendungen in verschiedenen Forschungsbereichen, einschließlich:
- RNA-Biologie ForschungUntersuchung der Rollen der 2'-O-Methylierung in der RNA-Funktion, Stabilität und der Regulation der Translation.
- KrankheitsforschungDie 2'-O-Methylierung ist mit verschiedenen Krankheiten verbunden (z. B. Krebs, neurodegenerative Erkrankungen). Sequenzierungstechnologie kann die Mechanismen dieser Modifikationen bei Krankheiten aufdecken.
- ArzneimittelentwicklungDie Identifizierung von Zielen im Zusammenhang mit 2'-O-Methylierung bildet eine Grundlage für die Entwicklung neuer Medikamente.
2'-O-RNA-Methylierungs-Sequenzierungs-Workflow
CD Genomics nutzt fortschrittliche Sequenzierungsplattformen und jahrelange Erfahrung, um umfassende 2'-O-RNA-Methylierungsnachweisdienste anzubieten. Diese Dienstleistungen umfassen hauptsächlich die Probenverarbeitung, Sequenzierung und anschließende bioinformatische Analyse.

Dienstspezifikationen
Musteranforderungen
|
|
 Klicken |
Sequenzierungsstrategie
|
 |
Bioinformatische Analyse
Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an:
|
Analyse-Pipeline
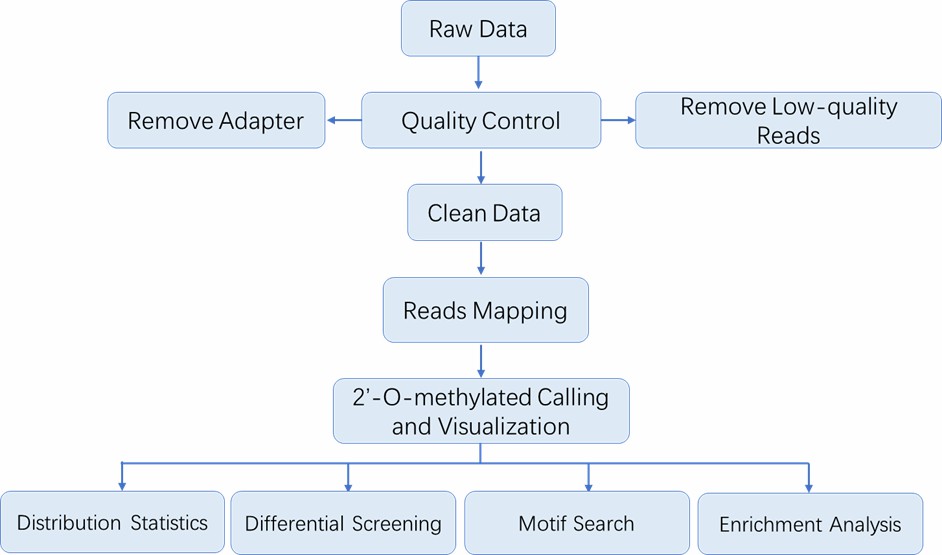
Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details zur 2'-O-RNA-Methylierungssequenzierung für Ihre Schrift (Anpassung)
Erforschen Sie RNA-Modifikationen mit den 2'-O-RNA-Methylierungssequenzierungsdiensten von CD Genomics. Wir bieten fortschrittliche Probenverarbeitung, präzise Sequenzierung und umfassende bioinformatische Analysen, um 2'-O-Methylierungsstellen zu identifizieren und zu quantifizieren. Kontaktieren Sie uns, um Ihre Forschung mit detaillierten Einblicken in die RNA-Methylierung zu verbessern.
Referenz
- Motorin Y, Marchand V. Nachweis und Analyse von RNA-Ribose-2'-O-Methylierungen: Herausforderungen und Lösungen. Gene, 2018, 9(12): 642.
Demonstrationsergebnisse
Teilweise Ergebnisse sind unten aufgeführt:
Referenz
- Dai Q, Moshitch-Moshkovitz S, Han D, et al. Nm-seq kartiert 2'-O-Methylierungsstellen in menschlicher mRNA mit Basengenauigkeit. Naturmethoden, 2017, 14(7): 695-698.
2'-O-RNA-Methylierung Seq FAQs
1. Warum ist rRNA an ausgewählten 2' OH methyliert?
rRNA (ribosomale RNA) unterliegt einer Methylierung an ausgewählten 2'-OH-Gruppen aus mehreren wichtigen Gründen:
Strukturelle StabilitätDie 2'-O-Methylierung trägt erheblich zur strukturellen Integrität der rRNA bei. Diese Modifikation spielt eine entscheidende Rolle bei der Erhaltung der korrekten dreidimensionalen Konformation der rRNA, die grundlegend für die ordnungsgemäße Assemblierung und Funktionalität des Ribosoms ist.
Widerstand gegen AbbauDie Methylierung an der 2'-OH-Position erhöht die Resistenz von rRNA gegenüber Ribonukleasen, Enzymen, die für den RNA-Abbau verantwortlich sind. Diese erhöhte Resistenz ist entscheidend für die Aufrechterhaltung der Langlebigkeit und Funktion von rRNA im Ribosom.
Genauigkeit der ÜbersetzungDie 2'-O-Methylierung ist entscheidend für die Genauigkeit der Proteinsynthese. Die modifizierten Nukleotide unterstützen die präzise Positionierung von tRNAs und mRNA während der Translation und minimieren somit Fehler in der Proteinsynthese.
Interaktion mit ribosomalen ProteinenMethylierte rRNA zeigt verbesserte Wechselwirkungen mit ribosomalen Proteinen und anderen translationsbezogenen Faktoren. Diese Wechselwirkungen sind entscheidend für die Zusammenstellung und Stabilität des ribosomalen Komplexes.
Funktionale BereicheDie Methylierung erfolgt an spezifischen Stellen, die für die ribosomale Aktivität entscheidend sind. Diese Modifikationen können die katalytischen Funktionen des Ribosoms und seine Wechselwirkungen mit anderen Molekülen beeinflussen, wodurch eine effiziente und effektive Proteinsynthese sichergestellt wird.
2. Welche Arten von RNA können auf 2'-O-Methylierung analysiert werden?
Dieses Sequenzierungsverfahren kann auf verschiedene RNA-Typen angewendet werden, einschließlich:
- Messenger-RNA (mRNA)
- Ribosomale RNA (rRNA)
- Transfer-RNA (tRNA)
- Kleine nukleäre RNA (snRNA)
- Lange nicht-codierende RNA (lncRNA)
3. Warum ist die 2'-O-RNA-Methylierung wichtig?
2'-O-RNA-Methylierung hat eine bedeutende Rolle für verschiedene zelluläre Prozesse, einschließlich:
- Verbesserung der RNA-Stabilität und Abbauresistenz: Diese Modifikation verleiht RNA-Molekülen erhöhte Stabilität und schützt sie vor dem Abbau durch Ribonukleasen und andere enzymatische Aktivitäten, die ansonsten die Integrität der RNA gefährden könnten.
- Erhaltung der richtigen RNA-Sekundär- und Tertiärstrukturen: Durch die Beeinflussung der Faltung und strukturellen Konformationen von RNA stellt die 2'-O-Methylierung sicher, dass RNA-Moleküle Konfigurationen annehmen, die für ihre biologischen Funktionen unerlässlich sind.
- Regulierung von RNA-Protein-Interaktionen: Diese Methylierung erleichtert spezifische und hochaffine Interaktionen zwischen RNA-Molekülen und RNA-bindenden Proteinen, die für verschiedene RNA-vermittelte Regulationsmechanismen von entscheidender Bedeutung sind.
- Genauigkeit und Effizienz in der Translation und Spleißung: 2'-O-Methylierung unterstützt die präzise und effektive Durchführung von Translation und Spleißprozessen, minimiert Fehler und verbessert die Gesamtfidelity und Effizienz der Proteinsynthese und RNA-Verarbeitung.
4. Wie bereite ich meine Proben für die 2'-O-RNA-Methylierungssequenzierung vor?
Um Proben vorzubereiten:
- Verwenden Sie frische oder ordnungsgemäß gelagerte Gewebe oder Zellen.
- Befolgen Sie die empfohlenen RNA-Extraktionsprotokolle, um hochqualitative RNA zu erhalten.
- Vermeiden Sie RNase-Kontamination, um RNA-Abbau zu verhindern.
5. Wie lange dauert der 2'-O-RNA-Methylierungs-Sequenzierungsprozess?
Die Bearbeitungszeit hängt von verschiedenen Faktoren ab, einschließlich der Anzahl der Proben, der Komplexität der Analyse und des Dienstleisters. In der Regel kann der Prozess von der Einreichung der Proben bis zur Datenlieferung mehrere Wochen in Anspruch nehmen.
2'-O-RNA-Methylierung Seq Fallstudien
Fallstudie 1:
Nm-seq kartiert 2'-O-Methylierungsstellen in menschlicher mRNA mit Basengenauigkeit.
Zeitschrift: Nature Methods
Impact-Faktor: 26,9
Veröffentlicht: 28. Februar 2018
Hintergrund
mRNA-Modifikationen, die Teil des Epitranskriptoms sind, regulieren die Genexpression und umfassen 2'-O-Methylierung (Nm). Nm ist in rRNA, tRNA, snRNA und Mikro-RNA reichlich vorhanden und für deren Funktion und Stabilität unerlässlich. Traditionelle Kartierungsmethoden, wie 2OMe-seq und RiboMeth-seq, identifizieren Nm-Stellen, indem sie Pausen in der reversen Transkription oder Widerstand gegen Hydrolyse nachweisen. Bestehende Methoden haben Schwierigkeiten mit seltener RNA oder Nm-Stellen mit niedriger Stöchiometrie. Die neue Nm-seq-Methode verwendet oxidative Spaltung, um Nm-Stellen mit hoher Sensitivität und Einzel-Nukleotid-Präzision zu kartieren.
Materialien & Methoden
Probenvorbereitung
- Menschliche HeLa (zervikales Adenokarzinom)
- HEK293 (humane embryonale Nieren) Zellen
- Totale RNA-Extraktion
- RNA-Reinigung
Sequenzierung
- Nm-seq
- Illumina HiSeq2500
- LC-MS/MS
- Identifizierung von Nm-Stellen in rRNA
- Identifizierung von Nm-Stellen im Transkriptom
- Motivsuche
- Metagenprofiling
- GO-Anreicherungsanalyse
- Statistische Analyse
Ergebnisse
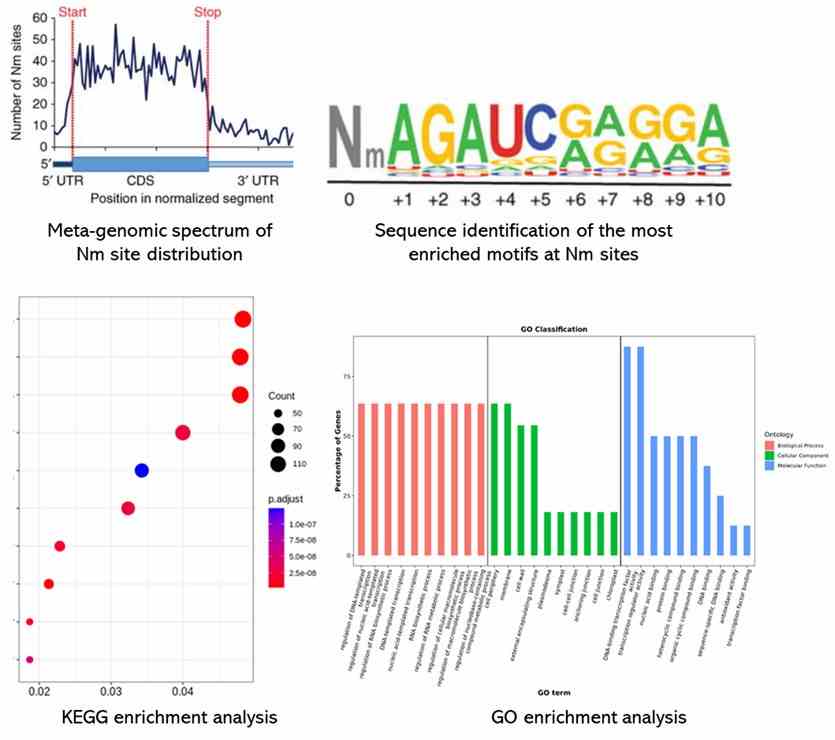
Dieser Artikel präsentiert zum ersten Mal die Identifizierung von Tausenden von Methylierungsstellen auf der mRNA menschlicher HeLa- und 293T-Zellen und beschreibt die Verteilung dieser Methylierungsstellen. Unter ihnen befinden sich die meisten Methylierungsstellen (95,7%) innerhalb von 2398 RefSeq-annotierten Genen, wobei 95,9% in proteincodierenden Genen auftreten und ein kleiner Teil in nicht-codierenden Genen (4,1%). Innerhalb der proteincodierenden Transkripte zeigt die Datenanalyse, dass die Mehrheit der Stellen im kodierenden Bereich (CDS) liegt (70,3%). Die Motivanalyse zeigt, dass die charakteristische Sequenz für 2'-O-Methylierung AGAU ist, gefolgt von einer hohen Verteilung von A- oder G-Basen. Darüber hinaus sind 2'-O-Methylierungsstellen in proteincodierenden Kodons für drei Aminosäuren angereichert.
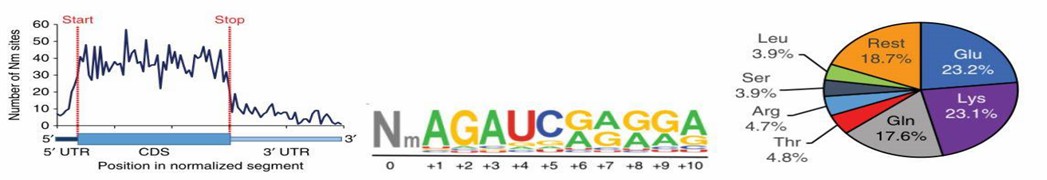 Abbildung 1: Eigenschaften von 2'-O-RNA-Methylierungsstellen auf menschlichen Zell-mRNA-Molekülen.
Abbildung 1: Eigenschaften von 2'-O-RNA-Methylierungsstellen auf menschlichen Zell-mRNA-Molekülen.
Fazit
Zusammenfassend zeigt diese Studie erstmals die Verteilungseigenschaften der 2'-O-Methylierung in menschlichen Zellen auf.
Fallstudie 2: Beteiligung der 2'-O-RNA-Methyltransferase FTSJ3 an der Regulation der angeborenen Immunität von HIV
Kürzlich entdeckte die von Professorin Yamina Bennasser geleitete Forschungsgruppe an der Universität Montpellier in Frankreich erstmals die Anwesenheit von 2'-O-Methylierungsstellen im HIV-Virus. Diese Stellen zeigen Aktivität bei der Infektion von Wirtszellen und werden von der Methyltransferase FTSJ3 reguliert.
FTSJ3 als 2'-O-Methyltransferase:
Durch Massenspektrometrie und Western-Blot-Experimente wurde beobachtet, dass das Methylierungsleseprotein TRBP mit der Methyltransferase FTSJ3 interagiert und den TRBP-FTSJ3-Komplex bildet.
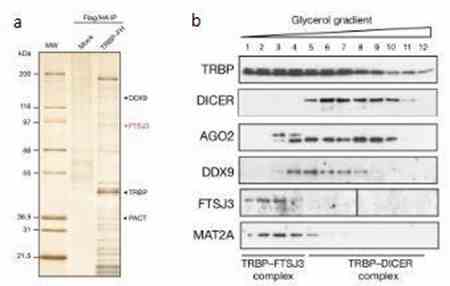 Abbildung 2: Bildung von zwei unterschiedlichen Komplexen durch TRBP.
Abbildung 2: Bildung von zwei unterschiedlichen Komplexen durch TRBP.
2'-O-Methylierung in HIV-1 RNA-Virionen:
Forscher verwendeten die 2'-O-Methylierung-Sequenzierung, um den Methylierungsstatus von HIV-1-RNA zu untersuchen. Sie identifizierten 17 2'-O-Methylierungsstellen, von denen 15 auf Adenin lagen. Die anschließende Methylierungssequenzierung von HIV-1, das in FTSJ3-Knockout-Zellen verpackt war, zeigte eine Verringerung der Methylierungsniveaus an mehreren Stellen. Dies weist auf den Einfluss der Methyltransferase FTSJ3 auf das 2'-O-Methylierungsmuster hin.
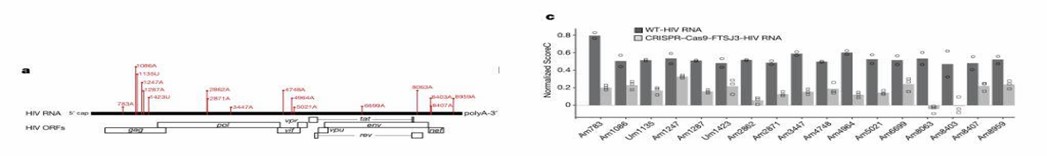 Abbildung 3: Methylierungsprofil von HIV-1 RNA und die Auswirkungen der Depletion von FTSJ3.
Abbildung 3: Methylierungsprofil von HIV-1 RNA und die Auswirkungen der Depletion von FTSJ3.
FTSJ3-Beteiligung am Immunregulationsmechanismus von HIV-1:
Auswirkung auf die IFN-α/IFN-β-Expression: Zunächst, beeinflusst intrazelluläres FTSJ3 die Infektion von Wirtszellen durch das Virus? Die Forscher verwendeten U937-monozytäre Zellen, die HIV-RNA exprimieren, mit entweder Wildtyp (WT) oder FTSJ3-siRNA-Knockdown. Die Ergebnisse zeigten einen signifikanten Anstieg der Expression von IFN-α/IFN-β in der Versuchsgruppe im Vergleich zur WT-Gruppe.
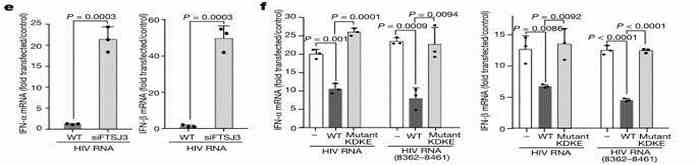 Abbildung 4: Beteiligung von FTSJ3 am Immunregulationsprozess von HIV-1.
Abbildung 4: Beteiligung von FTSJ3 am Immunregulationsprozess von HIV-1.
MDA5-Regulation von Methyltransferasen in der Immunmodulation von HIV-1:
MDA5, das als zytoplasmatischer RNA-Sensor fungiert, spielt eine entscheidende Rolle in der Immunantwort. Nach der Herabregulierung von FTSJ3 kam es zu einem Rückgang der IFN-Expression. Anschließend, nach der Herabregulierung von MDA5, nahm die IFN-Expression zu. Dieses Ergebnis bestätigt, dass die Methyltransferase zur Flucht der HIV-RNA vor der 2'-O-Methylierung durch MDA5 beiträgt und somit die Immunflucht vervollständigt.
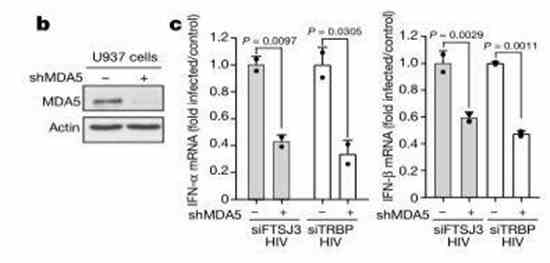 Abbildung 5: FTSJ3 moduliert die Empfindlichkeit des MDA5-IFN-Weges.
Abbildung 5: FTSJ3 moduliert die Empfindlichkeit des MDA5-IFN-Weges.
Die Depletion von FTSJ3 unterdrückt die HIV-Replikation.
Um die Auswirkungen von FTSJ3 auf die zelluläre Immunstimulation durch das Virus zu bewerten, verwendeten die Autoren FTSJ3-Knockout in MDDCs während der Virusinfektion. Offensichtlich induzierten FTSJ3-Knockout-Zellen die Expression der Typ-I-antiviralen Antwort auf HIV-Viruspartikel und hemmten gleichzeitig die Effizienz der HIV-1-Expression. Somit induziert siFTSJ3-HIV nicht-methylierte RNA die angeborene Immunität gegen virale RNA und unterdrückt die HIV-Replikation.
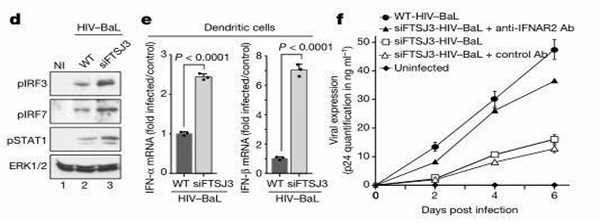 Abbildung 6: Depletion von FTSJ3 unterdrückt die HIV-Replikation.
Abbildung 6: Depletion von FTSJ3 unterdrückt die HIV-Replikation.
Zusammenfassend zeigt diese Studie erstmals die RNA 2'O-Methylierungsaktivität von FTSJ3 und bestätigt, dass das HIV-Virus das FTSJ3-TRBP-System nach der Infektion rekrutieren kann, um 2'O-Methylierungsmodifikationen an viraler RNA durchzuführen. Folglich verringert diese Modifikation die Empfindlichkeit des MDA5-IFN-Wegs, was die Erkennung durch das Immunsystem reduziert. Dieser neuartige Weg der HIV-1-Evasion des angeborenen Immunsystems bietet vielversprechende Ansätze als neues therapeutisches Ziel für die AIDS-Behandlung.
Referenzen
- Dai Q, Moshitch-Moshkovitz S, Han D, et al. Nm-seq kartiert 2'-O-Methylierungsstellen in menschlicher mRNA mit Basengenauigkeit. Naturmethoden, 2017, 14(7): 695-698.
- Ringeard, M., Marchand, V., Decroly, E. et al. FTSJ3 ist eine RNA 2'-O-Methyltransferase, die von HIV rekrutiert wird, um die Erkennung durch das angeborene Immunsystem zu vermeiden. Natur, 2019, 565, 500–504.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Die Spaltung von Phagen-DNA durch Restriktionsendonukleasen ermöglicht die Wiederbelebung aus der durch Cas13 induzierten bakteriellen Dormanz.
Zeitschrift: Nature Mikrobiologie
Jahr: 2023
IL-4 fördert die Erschöpfung von CD8.+ CART-Zellen
Zeitschrift: Nature Communications
Jahr: 2024
Fettreiche Diäten während der Schwangerschaft führen zu Veränderungen der DNA-Methylierung und Proteinexpression im Pankreasgewebe der Nachkommen: Ein Multi-Omics-Ansatz
Zeitschrift: Internationale Zeitschrift für Molekulare Wissenschaften
Jahr: 2024
KMT2A assoziiert mit dem PHF5A-PHF14-HMG20A-RAI1 Subkomplex in Stammzellen des Pankreaskarzinoms und reguliert epigenetisch deren Eigenschaften.
Zeitschrift: Naturkommunikationen
Jahr: 2023
Krebsassoziierte DNA-Hypermethylierung von Polycomb-Zielen erfordert die doppelte Erkennung von Histon H2AK119-Ubiquitinierung und der sauren Tasche des Nukleosoms durch DNMT3A.
Journal: Wissenschaftliche Fortschritte
Jahr: 2024
Genomisches Imprinting-ähnliches monoalleles väterliches Ausdrucksmuster bestimmt das Geschlecht von Kanalkatzenfischen.
Journal: Wissenschaftliche Fortschritte
Jahr: 2022
Mehr ansehen Artikel, die von unseren Kunden veröffentlicht wurden.


