
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Gezielte Regionssequenzierung
CD Genomics bietet seit Jahrzehnten einen schnellen und kostengünstigen Sequenzierungsdienst für gezielte Regionen an. Wir setzen modernste Sequenzierungsinstrumente ein, um Ihnen zu helfen, vollständige DNA-Informationen der ausgewählten Ziele auf kosteneffiziente Weise zu erhalten, was sowohl die Breite als auch die Tiefe Ihrer genomischen Forschung erheblich erhöhen kann.
Die Einführung der gezielten Regionssequenzierung
Die gezielte Regionssequenzierung ist ein effektiver Ansatz, um Ihre ausgewählten Interessensregionen zu untersuchen, indem Sie Next-Generation-SequenzierungDurch die Nutzung gezielter Regionensequenzierungs-Panels können Sie Einzel-Nukleotid-Polymorphismen (SNPs), Insertionen/Löschungen (InDels), Kopienzahlvariationen (CNVs) und strukturelle Varianten (SVs) entdecken. Im Vergleich zu Whole-Genome-SequenzierungDie gezielte Regionssequenzierung ermöglicht eine genaue Erkennung seltener Varianten mit höherer Sensitivität und Spezifität. Dieser Ansatz ist sehr kosteneffektiv, wenn eine große Anzahl von Proben bearbeitet wird, was die Kosten pro Probe erheblich senkt.
Der Prozess der gezielten Regionen-Sequenzierung umfasst die Gestaltung/Synthese von Sonden/Primern, die Erfassung der Zielregionen, den Aufbau der Bibliothek, die Paar-End-Sequenzierung und die bioinformatische Analyse basierend auf den Zielsequenzen. Spezifische Sonden-/Primer-Sets werden entworfen, um die gezielten Regionen entweder durch Hybridisierung oder Amplifikationsmethoden anzureichern. Bei der Hybridfängermethode werden biotinylierte Oligonukleotid-Sonden entworfen, um Zielregionen innerhalb einer DNA-Fragmentbibliothek anzusprechen. Nach einer Hybridisierungsinkubation werden streptavidin-beschichtete magnetische Perlen verwendet, um die biotinylierte Sonden/Ziel-Hybride zu erfassen, was zu einer Bibliothek führt, die stark angereichert ist für die gezielte DNA. Die aktuelle Amplifikationsmethode basiert auf Multiplex-PCR oder einer Form der Primerverlängerung über die interessierenden Regionen.
Die gezielte Regionen-Sequenzierung hat ein breites Anwendungsspektrum, einschließlich:
- Erkennung von SNPs/InDels/CNVs/SVs
- Entdeckung von Keimbahn- oder somatischen Mutationen
- Erkennung und Quantifizierung seltener Varianten und niederfrequenter Allele
- Verknüpfungsanalyse für erbliche Krankheiten
- Entdeckung von Biomarkern und therapeutischen Zielen
Vorteile der gezielten Regionssequenzierung
- Identifizierung von Varianten mit niedrigen Allelfrequenzen (bis zu 1%).
- Ein kleinerer Datensatz für die bioinformatische Analyse.
- Viel geringere Kosten bei einer großen Anzahl von Proben.
- Hohe Tiefe (500-1000X oder höher), die die Identifizierung seltener Varianten ermöglicht.
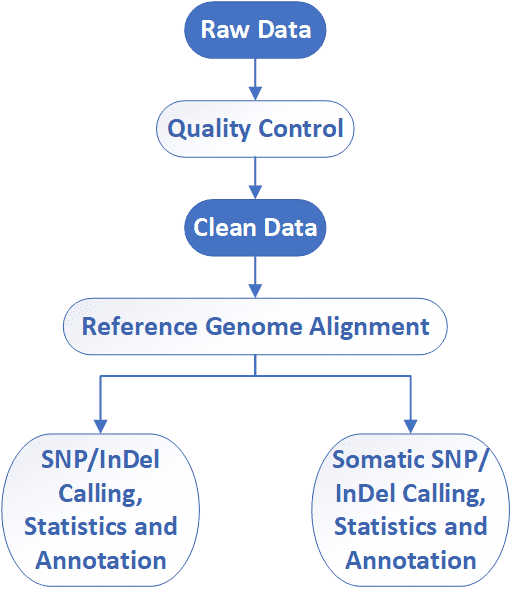
Zielgerichteter Regionensequenzierungs-Workflow
CD Genomics setzt Illumina HiSeq-Geräte ein, um eine schnelle und präzise Sequenzierung gezielter Regionen sowie bioinformatische Analysen anzubieten. Unsere hochqualifizierten Experten führen das Qualitätsmanagement durch und befolgen jeden Schritt, um hochwertige Ergebnisse zu gewährleisten. Der allgemeine Arbeitsablauf für die Sequenzierung gezielter Regionen ist unten skizziert.

Dienstspezifikationen
Beispielanforderungen
|
|
 Klicken |
Sequenzierungsstrategien
|
 |
Bioinformatikanalyse Wir bieten maßgeschneiderte bioinformatische Analysen an, einschließlich:
|
Analyse-Pipeline
Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details zur gezielten Regionssequenzierung für Ihre Schreibweise (Anpassung)
CD Genomics bietet ein umfassendes Paket für die Sequenzierung gezielter Regionen an, das die Standardisierung von Proben, das Design von Sonden/Primern, die gezielte Erfassung, den Bibliotheksaufbau, die Tiefensequenzierung, die Qualitätskontrolle der Rohdaten und die bioinformatische Analyse umfasst. Wir können diese Pipeline an Ihr Forschungsinteresse anpassen. Wenn Sie zusätzliche Anforderungen oder Fragen haben, zögern Sie bitte nicht, uns zu kontaktieren.
Demo-Ergebnisse

Häufig gestellte Fragen zu Targeted Region Seq
1. Was ist der Unterschied zwischen Whole Exome Sequencing und gezieltem Regionssequencing?
Trotz wahrscheinlich desselben Prinzips und Arbeitsablaufs, Whole-Exom-Sequenzierung fokussiert sich auf das Exom, während die gezielte Regionssequenzierung sich auf alle Gene konzentriert, die von Ihnen definiert werden.
2. Was muss ich einreichen?
Der Kunde, der den Service für die gezielte Region benötigt, muss die DNA- oder Gewebeproben sowie die Liste der Genkandidaten oder die chromosomale Lokalisation der Zielregion einreichen. Wir sind verantwortlich für die Generierung der Sonden, die Anreicherung des Ziels, den Bibliotheksaufbau, die Sequenzierung und die bioinformatische Analyse.
Wir akzeptieren die folgenden Probenarten für die gezielte Regionen-Sequenzierung: gereinigte genomische DNA, PCR-Amplikate, gefrorene Zellpellets, Bakterienkolonien, FFPE (formalin-fixiertes, paraffin-eingebettetes) Gewebe, Gewebe, Blut und Abstriche.
3. Wie sieht es mit der Bearbeitungszeit aus?
Die gezielte Regionssequenzierung erfordert maßgeschneiderte Sonden, und die maßgeschneiderte Sonde muss weiter optimiert werden, um eine effektive Anreicherung der Zielregionen zu gewährleisten. Es dauert oft 40 Arbeitstage, um die Anpassung der Sonden und die Tiefensequenzierung abzuschließen. Es gibt jedoch kommerzielle Lösungen für die gezielte Regionssequenzierung, wie die SeqCap EZ Prime Choice Sonden, die den Prozess der Anpassung der Sonden eliminieren können.
4. Wie validiert man Varianten?
Identifizierte Varianten durch die Next-Generation-Sequenzierung (NGS) benötigen weitere Bestätigung mittels entweder NGS oder Sanger-SequenzierungLaut der Umfrage von Coppieters im September 2014 gaben fast 70 % der 178 Befragten an, dass sie derzeit ihre NGS-Ergebnisse mit NGS- oder Sanger-Sequenzierung validieren. Wir werden Varianten mit NGS und Sanger-Sequenzierung validieren.
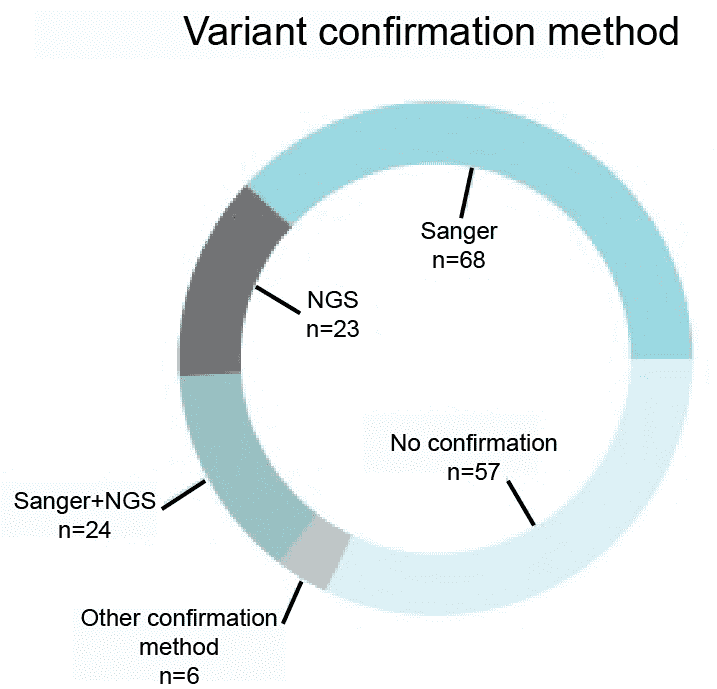 Abbildung 1. Eine Umfrage zu den Methoden zur Bestätigung von Varianten (Coppieters et al. 2016).
Abbildung 1. Eine Umfrage zu den Methoden zur Bestätigung von Varianten (Coppieters et al. 2016).
Referenz:
- Coppieters F, Verniers K, De Leeneer K, et al. Zielgerichtete Nachsequenzierung und Variantenvalidierung mit pxlence PCR-Assays. Biomolekulare Detektion und Quantifizierung, 2016, 6: 22-26.
Fallstudien zur gezielten Regionensequenzierung
Identifizierung einer neuartigen missense Mutation von MIP in einer chinesischen Familie mit kongenitalen Katarakten durch gezielte Regionen-Sequenzierung
Journal: Wissenschaftliche Berichte
Impactfaktor: 4,259
Veröffentlicht: 06. Januar 2017
Zusammenfassung
Die Autoren rekrutierten eine chinesische Familie mit autosomal dominanten kongenitalen Katarakten (ADCC). Sie fanden eine heterozygote Missense-Mutation c.634G>C (p.G212R) Substitution in der MIP Gen durch gezielte Regionssequenzierung. Zusammenfassend präsentiert die Studie genetische und funktionale Beweise für diese neuartige Mutation, die zu kongenitalen progressiven kortikalen Punktierungen mit oder ohne Y-Nähten führt. Der Proband (II:2) hatte einen anderen Typ von
Ergebnisse
1. Klinische Merkmale
Die Autoren untersuchten eine chinesische Familie (Abbildung 1) und stellten fest, dass der Proband (II:2) eine punktuelle Katarakt aufwies, während die anderen betroffenen Mitglieder punktuelle kortikale Trübungen in Kombination mit Y-suturalen Katarakten zeigten (Abbildung 2).
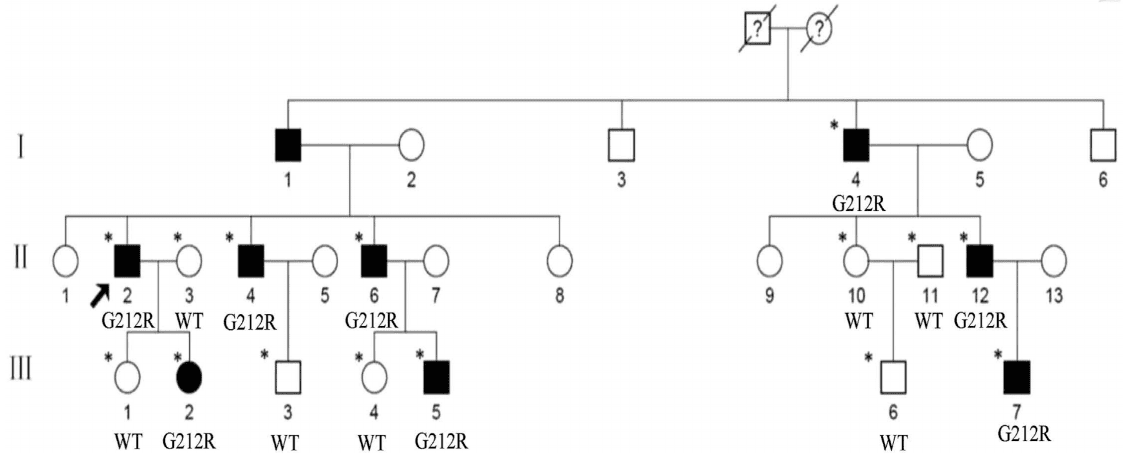 Abbildung 1. Stammbaum der Familie. Quadrate und Kreise stehen für Männer und Frauen. Schwarze Symbole kennzeichnen betroffene Mitglieder, während offene Symbole unbeeinträchtigte Personen darstellen. Die diagonale Linie kennzeichnet ein verstorbenes Familienmitglied, und der schwarze Pfeil zeigt den Probanden an. Sterne kennzeichnen sequenzierte Individuen.
Abbildung 1. Stammbaum der Familie. Quadrate und Kreise stehen für Männer und Frauen. Schwarze Symbole kennzeichnen betroffene Mitglieder, während offene Symbole unbeeinträchtigte Personen darstellen. Die diagonale Linie kennzeichnet ein verstorbenes Familienmitglied, und der schwarze Pfeil zeigt den Probanden an. Sterne kennzeichnen sequenzierte Individuen.
 Abbildung 2. Spaltlampenfotografien der Patienten. Der Proband II:2 (A) zeigte einen punktuellen Katarakt, während sein jüngerer Bruder II:6 (B) und der Sohn des Bruders III:5 (C) einen Y-Nähtenkatarakt in Kombination mit punktuellen kortikalen Opazitäten zeigten.
Abbildung 2. Spaltlampenfotografien der Patienten. Der Proband II:2 (A) zeigte einen punktuellen Katarakt, während sein jüngerer Bruder II:6 (B) und der Sohn des Bruders III:5 (C) einen Y-Nähtenkatarakt in Kombination mit punktuellen kortikalen Opazitäten zeigten.
2. Zielregionensequenzierung und Variantenanalyse
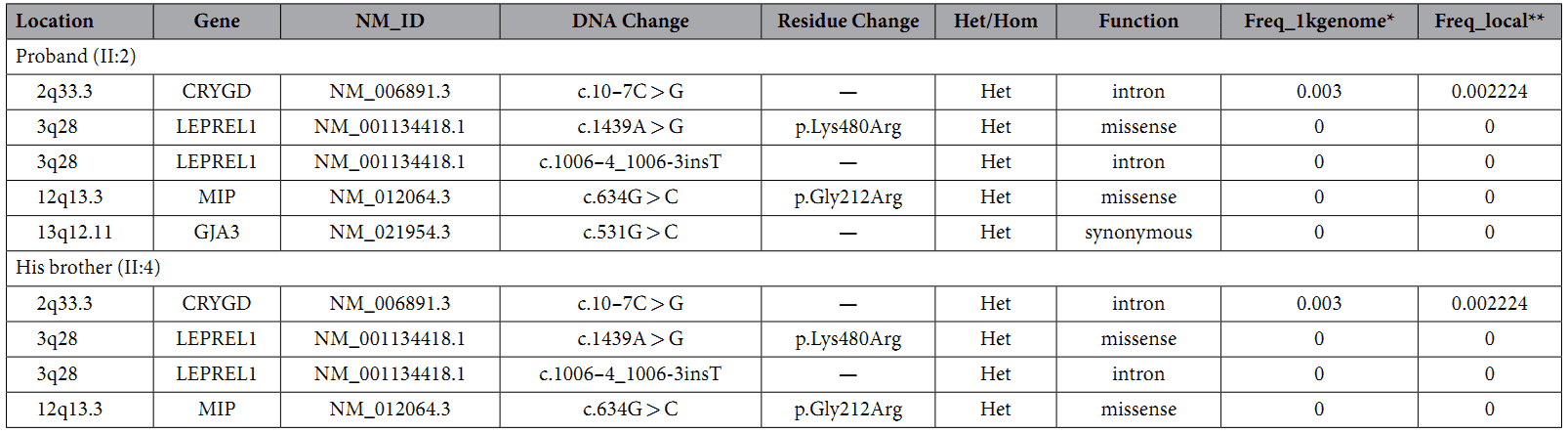
Ein maßgeschneidertes Erfassungsfeld wurde entworfen, um 351 Gene für genetische Augenerkrankungen zu erfassen, die aus OMIM (Human Genetics Knowledge for the World) und der veröffentlichten Literatur gesammelt wurden. Die Autoren fanden vier seltene Varianten in beiden Proben (Tabelle 1).
Tabelle 1. Seltene Varianten in Genen, die kongenitale Katarakte verursachen.
3. c.634G>C Mutation
Nur die c.634G>C-Mutation des MIP Das Gen war eine neuartige heterozygote Missense-Mutation, die Glycin an Position 212 in Arginin (p.G212R) umwandelte. Die Segregationsanalyse ergab, dass die Mutation gut mit allen betroffenen Teilnehmern cosegregierte und bei nicht betroffenen Mitgliedern oder den 100 normalen Kontrollen nicht nachgewiesen wurde.
PolyPhen-2 und SIFT-Analysen deuteten stark darauf hin, dass die p.G212R-Mutation wahrscheinlich schädlich für das Protein ist und möglicherweise für die kongenitalen Katarakte verantwortlich ist. Eine multiple Sequenzalignment zeigte, dass das Glycin an Position 212 von MIP ist unter verschiedenen Arten stark konserviert (Abbildung 3B).
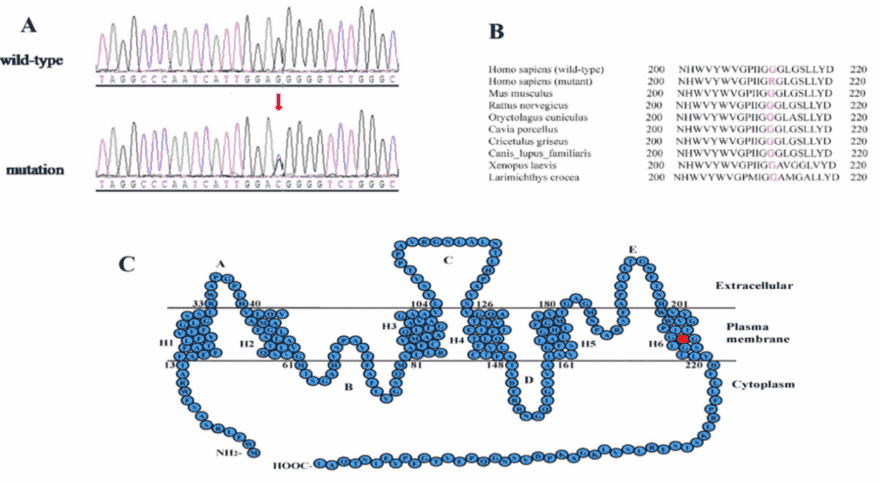 Abbildung 3. Mutationsanalyse des MIP Gen.
Abbildung 3. Mutationsanalyse des MIP Gen.
mRNA- und Proteinspiegel von WT-MIP und G212R-MIP
Die Autoren untersuchten die Expression des Wildtyps. MIP und G212R-MIP in transfizierten Hela-Zellen. Es zeigte sich, dass die grüne Fluoreszenz in Hela-Zellen, die mit dem G212R-Konstrukt transfiziert wurden, viel schwächer war als die in Zellen, die mit dem Wildtyp-Konstrukt transfiziert wurden.
Die RT-PCR wurde verwendet, um das RNA-Transkriptionsniveau von WT- zu messen.MIP und G212R-MIP, was revealed, dass ein ähnlicher Verwandter MIP mRNA-Expressionsniveaus (Abbildung 4B). Dennoch zeigte der Western Blot, dass die Proteinexpressionsniveaus von MIP wurden signifikant im G212R- reduziertMIP Zellen (Abbildung 4C) stimmen mit den Ergebnissen der Fluoreszenzmikroskopie-Analyse überein.
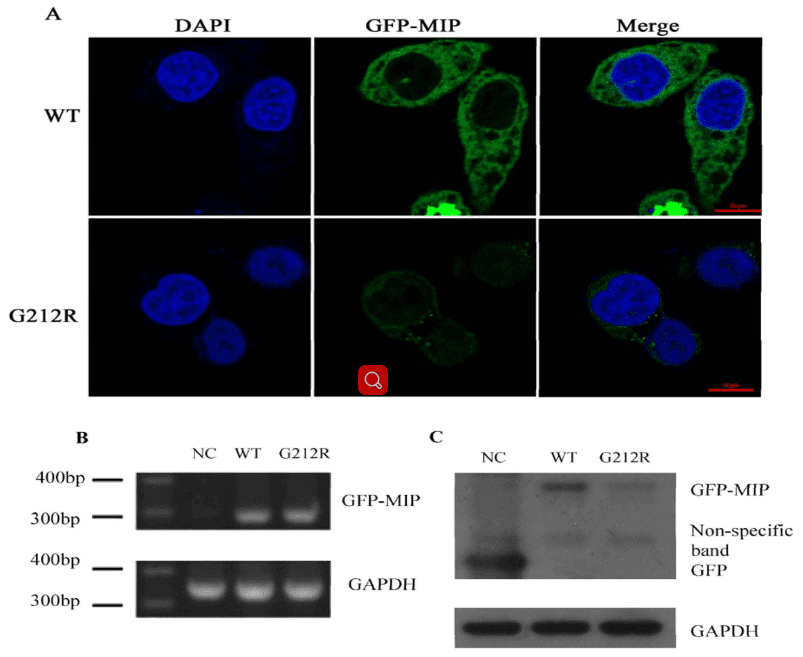 Abbildung 4. mRNA- und Proteinspiegel von WT-MIP und G212R-MIP.
Abbildung 4. mRNA- und Proteinspiegel von WT-MIP und G212R-MIP.
Schlussfolgerungen
In der vorliegenden Studie schlagen die Autoren vor, dass die c.634G>C, G212R Substitution zur Pathogenese von ADCC in dieser chinesischen Familie beitragen könnte.
Die Zielregion-Sequenzierung ist ein leistungsfähiges Werkzeug für die klinische Diagnose einer heterogenen Gruppe von monogenen Erkrankungen, aufgrund der kompatiblen Leistung bei der Erfassungsabdeckung, der niedrigeren Kosten und der kürzeren Bearbeitungszeit.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Genetische Varianten im Risikolokus SYNE1 für bipolare Störung, die die CPG2-Expression und Proteinfunktion beeinflussen.
Journal: Molekulare Psychiatrie
Jahr: 2019
Hochdichte-Kartierung und Kandidatengenanalyse von Pl18 und Pl20 in Sonnenblumen durch Whole-Genome-Resequenzierung
Internationales Journal für Molekulare Wissenschaften
Jahr: 2020
Identifizierung von Faktoren, die für die m6A mRNA-Methylierung in Arabidopsis erforderlich sind, zeigt eine Rolle für die konservierte E3-Ubiquitin-Ligase HAKAI.
Zeitschrift: New Phytologist
Jahr: 2017
Generierung eines hoch attenuierten Stammes von Pseudomonas aeruginosa für die kommerzielle Produktion von Alginat
Journal: Mikrobielle Biotechnologie
Jahr: 2019
Kombinationen von Bakteriophagen sind wirksam gegen multiresistente Pseudomonas aeruginosa und erhöhen die Empfindlichkeit gegenüber Carbapenem-Antibiotika.
Journal: Viren
Jahr: 2024
Genom-Analyse und Replikationsstudien des afrikanischen Grünen Affen Simian Foamy Virus Serotyp 3 Stamm FV2014
Journal: Viren
Jahr: 2020
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


