
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Virales Genom-Sequenzierung
CD Genomics bietet einen Service zur Sequenzierung von Virusgenomen auf den Plattformen Illumina und PacBio an. Wir können hochwertige von Neuem Zusammenstellung großer viraler Genome und höchste mögliche Datenqualität zu niedrigen Kosten.
Was ist die virale Genomsequenzierung?
Ein Virus besteht entweder aus einem einzelnen Nukleinsäuremolekül (DNA oder RNA), das von Proteinen umschlossen ist, oder einfach nur aus Proteinen, wie es beim Prionvirus der Fall ist. Die Klassifizierung von Viren kann auf der Zusammensetzung ihres genetischen Materials basieren, das in einzelsträngige DNA-Viren (ssDNA), doppelsträngige DNA-Viren (dsDNA), einzelsträngige RNA-Viren (ssRNA), doppelsträngige RNA-Viren (dsRNA) und Proteinviren wie das Prionvirus unterteilt wird. Darüber hinaus beeinflusst auch der Wirtstyp die Kategorisierung, was zu Differenzierungen wie Bakteriophagen (bakterielle Viren), Pflanzenviren (zum Beispiel das Tabakmosaikvirus) und Tierviren (einschließlich des Vogelgrippevirus, des Pockenvirus, HIV und anderer) führt.
Die virale Genomsequenzierung stellt eine entscheidende Technik dar, die in der modernen Virologie genutzt wird und auf den Hochdurchsatz-Sequenzierungstechnologien der zweiten Generation basiert. Sie umfasst das Erwerben von viralen Genomsequenzen sowie die Interpretation der kodierten Informationen und Variantendaten durch Bioinformatik Analytics. Eine häufig genutzte Plattform für die Next-Generation-Sequenzierung (NGS) ist Illumina, die eine Analyse von maximalen Fragmentlängen von etwa 300-500 Nukleotiden ermöglicht. Kürzlich wurden Fortschritte in der Sequenzierungstechnologie erzielt durch Der Aufstieg von PacBio mit der Einzelmolekül-Realtime (SMRT) SequenzierungHier sind vollständige Long-Reads mit minimalen Fehlern erreichbar, wodurch einige der Einschränkungen umgangen werden, die mit traditionellen NGS-Methoden verbunden sind.
Die CD-Genomik-Plattform birgt großes Potenzial für die Sequenzierung von Virusgenomen. Umfassende Virussequenzen werden die Interpretation von virale Metagenomik Daten durch Bereitstellung von Referenzgenomen führen zu einem besseren Verständnis der Virusvielfalt, Ökologie, Anpassung und Evolution und ermöglichen die Vorhersage von aufkommenden Infektionskrankheiten, die durch Viren verursacht werden.
Vorteile unseres Virusgenom-Sequenzierungsdienstes
- Keine Referenzsequenz erforderlich
- Sequenzen mit erheblicher Tiefe machen für Ihr Projekt Sinn.
- Vollständige Charakterisierung von viralen Genomen
- Identifizieren und quantifizieren Sie minor Varianten.
- Vollständig generieren von neuem Assemblierungen großer viraler Genome
- Kosteneffiziente Hochdurchsatz-Sequenzierung
- Hochwertige Daten und schnelle Bearbeitung
Anwendung der viralen Genomsequenzierung
- Studie über virale Evolution und Übertragungsdynamik
- Impfstoffdesign und Wirksamkeitsüberwachung
- Überwachung von Arzneimittelresistenzen
- Krankheitsdiagnose und -screening
- Studie der Wirt-Virus-Interaktionen
- Umweltüberwachung und Virusverfolgung
- Gentechnik und virale Therapien
Viralgenom-Sequenzierungs-Workflow
Dienstspezifikationen
Beispielanforderungen
|
|
 |
Sequenzierung
|
 |
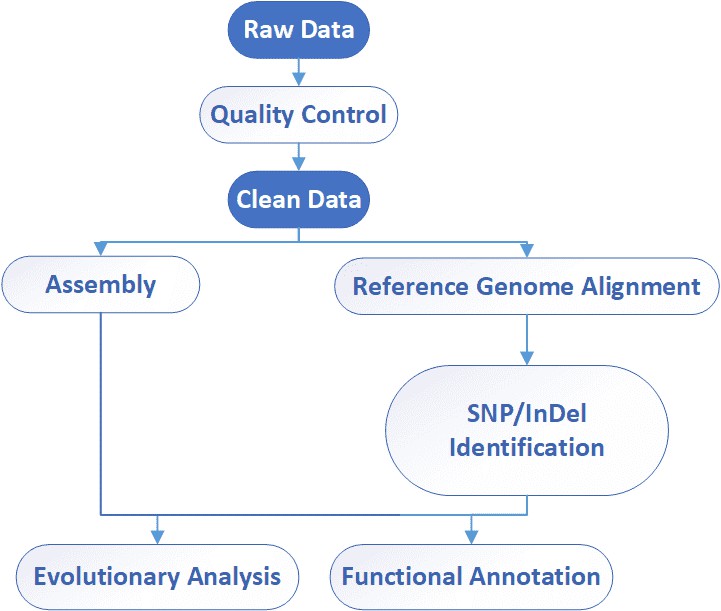
Bioinformatikanalyse
|
Analyse-Pipeline
Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details zur viralen Genomsequenzierung für Ihre Schreibanpassung.
Die virale Genomsequenzierung ist eine schnelle und effiziente Methode für die Forschung zur viralen Replikation, Verpackung, Funktion der Terminase, Transkriptionsregulation und dem Stoffwechsel der Wirtszelle. CD Genomics kann hochwertige Sequenzierungsdaten für Ihr Virusgenom von Interesse liefern. Kontaktieren Sie uns für Unterstützung bei der Konfiguration Ihres Projekts.
Referenzen:
- Houldcroft C J, Beale M A, Breuer J. Klinische und biologische Erkenntnisse aus der viralen Genomsequenzierung. Nature Reviews Mikrobiologie, 2017, 15(3): 183-192.
- Marston D A, McElhinney L M, Ellis R J, et al. Next-Generation-Sequenzierung von viralen RNA-Genomen. BMC Genomics, 2013, 14: 1-12.
Häufig gestellte Fragen zur viralen Genomsequenzierung
1. Welche Methoden zur Sequenzierung von viralen Genomen gibt es?
Genomsequenzierungstechniken, die regelmäßig für die Analyse von Viren verwendet werden, umfassen Whole Genome Sequencing (WGS), Gezielte Sequenzierung, Metagenomische Sequenzierung, MetatranskriptomikJede Methode weist ihre eigenen Vor- und Nachteile auf und kann selektiv basierend auf dem spezifischen Forschungsziel und dem empirischen Kontext ausgewählt werden.
2. Wie werden Sequenzdaten während der viralen Genomsequenzierung verarbeitet?
Die aus der Sequenzierung viraler Genome generierten Sequenzdaten erfordern typischerweise Analyse-Schritte wie Qualitätskontrolle, Sequenzanpassung, Variationsdetektion und funktionale Annotation. Zu den Prozessen der Qualitätskontrolle gehört die Entfernung von Sequenzen niedriger Qualität und Sequenzkontamination, um die Zuverlässigkeit der Daten sicherzustellen. Anschließend müssen die Sequenzdaten mit bekannten viralen Genomsequenzen verglichen werden, um Variantenpunkte innerhalb der Sequenz zu identifizieren und diese zu annotieren, was zu einem Verständnis potenzieller Funktionen und Pathogenität beiträgt.
3. Welche Bedeutung hat die Sequenzierung des Virusgenoms für Epidemien?
Durch die Sequenzierungsanalyse von Virusgenomen hilft uns, den Ursprung und den Übertragungsweg von Viren nachzuvollziehen, den Beginn von Krankheitsausbrüchen zu identifizieren und die Wege der viralen Verbreitung sowie deren Umfang zu verstehen. Dieser Ansatz unterstützt die Umsetzung zeitnaher Kontrollmaßnahmen und Notfallreaktionen, wodurch die Ausbreitung und Vermehrung der Epidemie effektiv eingedämmt wird, was die öffentliche Gesundheit und die gesellschaftliche Stabilität schützt.
Die konsistente Sequenzierung von Virusgenomen, die Erkennung mutationaler Veränderungen innerhalb des Virus, erleichtert das Studium der sequenzbasierten Virus-Evolution und identifiziert schnell aufkommende Variantenstämme. Dies bietet ein präventives Warnsystem und liefert entscheidende Hinweise für das Management von Krankheitsausbrüchen. Anschließend bildet diese wertvolle Information eine entscheidende Referenz für die Gestaltung und Entwicklung von Impfstoffen.
Fallstudien zur viralen Genomsequenzierung
Sequenzierung viraler Genome aus einer einzelnen isolierten Plaque
Journal: Virologie Journal
Impactfaktor: 5,913
Veröffentlicht: 06. Juni 2013
Hintergründe
Die Sequenzierung von viralen und Bakteriophagen-Genomen stößt häufig auf Hindernisse aufgrund des erheblichen Bedarfs an genomischem Material. Die vorliegende Studie beschreibt eine Methode, die die Einzelplaque-Sequenzierung mit der sequenzunabhängigen Amplifikation mit einem einzelnen Primer (SISPA) optimiert. Diese Methodik ist anwendbar für die Next-Generation-Sequenzierung des gesamten Genoms von jedem kultivierbaren Virus, wodurch die Notwendigkeit für die großangelegte Produktion von Virusbeständen oder die Anwendung von Zentrifugationstechniken zur Virusreinigung entfällt.
Methoden
- Influenza-Virus-Plaque-Wachstum
- Nukleinsäureextraktion
- Verstärkung von doppelsträngigen SISPA-Produkten
- 454-Sequenzierung
- Illumina-Sequenzierung
- Sequenzvorverarbeitung
- Sequenzassemblierung und -zuordnung
- Identifizierung von SNPs
Ergebnisse
Die Sequenzdatenanalyse der Studie zeigt, dass aus einem einzelnen Plaque von Influenza A ein Mapping-Assembly mit einer durchschnittlichen Abdeckung von 3590-fach, das 100% des Genoms repräsentiert, erzeugt werden kann. De novo zusammengestellte Daten erstellen eine Überlappungsgruppe mit einer durchschnittlichen Sequenzabdeckung von 30-fach, die 96,5% des Genoms abdeckt. Im SISPA-Experimentalschema ergab nur die Sequenzierung von 10 pg Ausgangs-DNA von Phage F_HA0480sp/Pa1651 eine Mapping-Assemblierung mit einer durchschnittlichen Sequenzabdeckung von 3488-fach, was 99,9% des Referenzgenoms entspricht, sowie von neuem Assemblierung mit einer durchschnittlichen Sequenzabdeckungsrate von 45-fach, die 98,1% des Genoms abdeckt.
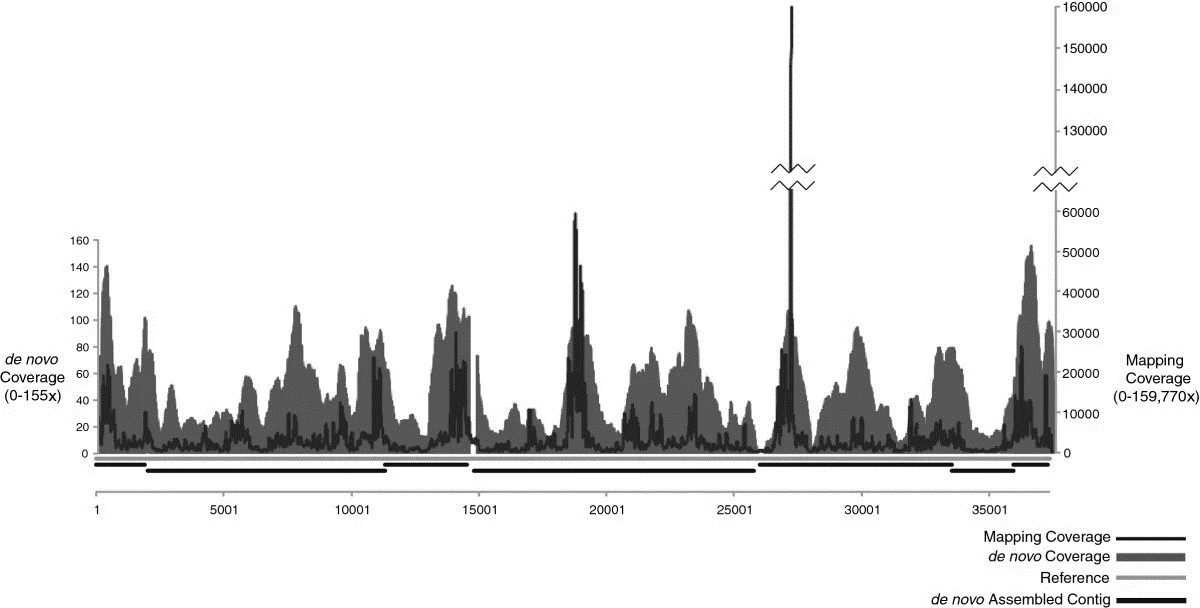 Abbildung 1. Kartierung und von Neuem Versammlung der Abdeckungssequenzierungsergebnisse für das Phagen-SISPA-Produkt aus 10 pg genomischer DNA.
Abbildung 1. Kartierung und von Neuem Versammlung der Abdeckungssequenzierungsergebnisse für das Phagen-SISPA-Produkt aus 10 pg genomischer DNA.
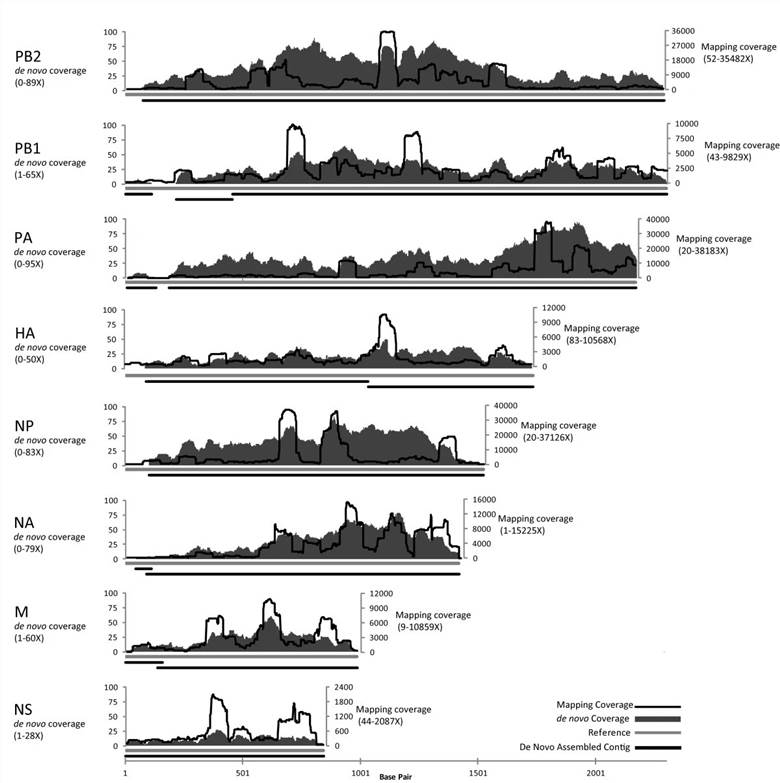 Abbildung 2. Kartierung und von Neuem Versammlung Abdeckungssequenzierungsergebnisse für das Influenza SISPA-Produkt aus einer einzelnen Plaque.
Abbildung 2. Kartierung und von Neuem Versammlung Abdeckungssequenzierungsergebnisse für das Influenza SISPA-Produkt aus einer einzelnen Plaque.
Fazit
Das Papier erläutert ein optimiertes Sequenzunabhängiges, Einzelprimer-Amplifikationsprotokoll (SISPA), das in der Lage ist, amplifizierte Produkte zu erzeugen. Diese tragen wertvolle hochqualitative Daten bei für von neuem Die Assemblierung während der Sequenzierung. Der Ansatz erfordert lediglich einen einzelnen Virusplaque oder eine minimale Vorlage von 10 pg DNA, was die schnelle Identifizierung von Viren während Ausbrüchen und solchen mit begrenzter Übertragung erleichtert. Dieser Aspekt unterstreicht das Potenzial der Methode, Herausforderungen bei der Virusdetektion und -kontrolle zu bewältigen.
Referenz:
- DePew J, Zhou B, McCorrison J M, et al. Sequenzierung viraler Genome aus einem einzelnen isolierten Plaque. Virologie-Zeitschrift, 2013, 10: 1-7.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Genomanalysen und Replikationsstudien des afrikanischen grünen Affen Simian Foamy Virus Serotyp 3 Stamm FV2014
Journal: Viren
Jahr: 2020
Vollständige Genomsequenz eines natürlich vorkommenden simianischen Schaumvirus-Isolats von Rhesusmakaken (SFVmmu_K3T)
Journal: Genom-Ankündigungen
Jahr: 2017
Identifizierung von Faktoren, die für die m6A mRNA-Methylierung in Arabidopsis erforderlich sind, zeigt eine Rolle für die konservierte E3-Ubiquitin-Ligase HAKAI.
Zeitschrift: New Phytologist
Jahr: 2017
Generierung eines hoch attenuierten Stammes von Pseudomonas aeruginosa für die kommerzielle Produktion von Alginate
Journal: Mikrobielle Biotechnologie
Jahr: 2019
Kombinationen von Bakteriophagen sind wirksam gegen multiresistente Pseudomonas aeruginosa und erhöhen die Empfindlichkeit gegenüber Carbapenem-Antibiotika.
Journal: Viren
Jahr: 2024
Genetische Varianten im Risikolokus SYNE1 für bipolare Störung, die die CPG2-Expression und Proteinfunktion beeinflussen.
Zeitschrift: Molekulare Psychiatrie
Jahr: 2019
Mehr ansehen Artikel, die von unseren Kunden veröffentlicht wurden.


