
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Mikrobielle Gesamte Genom-Sequenzierung
Mit jahrzehntelanger Erfahrung in den Bereichen von GenomsequenzierungCD Genomics widmet sich der Bereitstellung eines genauen und erschwinglichen Mikrobiom-Ganzgenom-Sequenzierungsdienstes. Wir kombinieren sowohl Illumina (kurze Reads) als auch PacBio (Langzeitleser) Plattformen für mikrobielle Neusequenzierung und vollständiges Genom von neuem SequenzierungUnsere starke Expertise wird durch flexible Sequenzierungsstrategien und Professionalität ergänzt. Bioinformatik Pipelines.
Die Einführung der mikrobiellen Gesamtengenomsequenzierung
Es gibt große Unterschiede zwischen Mikroorganismen und höheren Eukaryoten. Neben ihrem kleineren Genom haben die meisten Bakterien ein einzelnes zirkuläres Chromosom (manchmal mehr als ein Chromosom, lineare Chromosomen oder Kombinationen aus linearen und zirkulären Chromosomen). Die Gene in mikrobiellen Genomen sind normalerweise ein einzelner kontinuierlicher DNA-Strang, obwohl mehrere Arten von Introns selten im bakteriellen Genom vorkommen. Das Vorhandensein von Plasmiden im bakteriellen Genom ist ein weiterer wesentlicher Unterschied. Plasmide, die zirkuläre extrachromosomale DNA sind, können durch horizontalen DNA-Transfer übertragen werden, was die schnelle Evolution von Mikroorganismen vermittelt. Ein Virus ist ein nicht-zellulärer infektiöser Organismus, der aus einem Kern von DNA oder RNA besteht, der von einer Proteinhülle umgeben ist.
Die mikrobielle Ganzgenomsequenzierung liefert eine Fülle von Daten, die eine umfassende Bewertung aller genetischen Merkmale eines isolierten Mikroorganismus ermöglichen. Shotgun-Sequenzierung Strategie ist eine primäre Methode der mikrobiellen Ganzgenomsequenzierung. Die Sequenzierungsschritte erfordern kein arbeitsintensives Mapping und Klonierung, was enorme Zeit und Kosten spart. Darüber hinaus, Hochdurchsatz-Sequenzierung ermöglicht es uns, Hunderte von Bakterien oder Viren gleichzeitig mit der Kraft des Multiplexings zu sequenzieren. Bei der Whole-Genome-Shotgun-Sequenzierung wird das gesamte Genom in kleine Fragmente zerlegt, die sequenziert werden, und dann mithilfe von computergestützten Methoden basierend auf den überlappenden Regionen wieder zusammengesetzt, wodurch kein Referenzgenom erforderlich ist. PacBio SMRT-Technologie ermöglicht uns, bereitzustellen bakteriell von neuem Whole-Genome-Sequenzierung und pilzartig von Neuem Whole-Genome-Sequenzierung die genauere und zusammenhängendere Sequenzen erzeugen.
Die mikrobielle Ganzgenomsequenzierung ist entscheidend für die präzise mikrobielle Identifizierung und die Erstellung vollständiger Referenzgenome.von Neuem Sequenzierung), vergleichende genomische Studien (Re-Sequenzierung) und genomische Ausbeutung. Vergleichende genomische Studien können individuelle genetische Variationen und großflächige strukturelle Variationen innerhalb einer Population identifizieren, für die ein Referenzgenom verfügbar ist. Evolutionäre Merkmale und phylogenetische Beziehungen können daher abgeleitet werden. Die vollständige Mikrogenomsequenzierung bietet die Möglichkeit der Genfindung und -annotation. Nachdem mehrere Gene erklärt wurden, werden wahrscheinlich neuartige biochemische Wege identifiziert, die für die Medizin und Biotechnologie von Nutzen sein könnten.
Vorteile der mikrobiellen Ganzgenomsequenzierung
- Ein zeiteffektiver und kosteneffizienter Ansatz
- Breite Anwendungen: von neuem Sequenzierung, Genannotationen, vergleichende genomische Studien, evolutionäre Studien von Mikroorganismen usw.
- Arzneimittelentdeckung und -entwicklung: Bewerten Sie den Beitrag von DNA zur Pathogenese und verstehen Sie die Rolle mobiler Elemente bei Arzneimittelresistenz und Übertragung.
Anwendung der mikrobiellen Gesamten Genomsequenzierung
- Pathogenidentifikation: Schnelle und präzise Bestimmung der Pathogenidentität durch den Vergleich mit etablierten Genomdatenbanken, die klinische Diagnosen und epidemiologische Untersuchungen erleichtert.
- Überwachung der Antibiotikaresistenz: Identifizierung und Analyse von Resistenzgenen zur Aufdeckung mikrobieller Resistenzmechanismen, die Anleitung für das klinische Management und die Überwachung der Verbreitung von Resistenzen bieten.
- Genomvergleiche: Vergleichende Analyse von Genomen verschiedener Stämme zur Abgrenzung genetischer Variationen, die das Verständnis von mikrobieller Evolution, genetischer Vielfalt und Populationsdynamik unterstützt.
- Funktionelle Genomik: Entdeckung funktioneller Gene und Stoffwechselwege innerhalb von Genomen zur Entschlüsselung mikrobiologischer Eigenschaften und ökologischer Rollen.
- Umweltverschmutzungsüberwachung: Sequenzierungsanalyse von mikrobiellen Gemeinschaften in der Umwelt zur Überwachung der Auswirkungen von Schadstoffen und mikrobiellen Reaktionen, Bewertung der ökologischen Integrität.
- Arzneimittelentdeckung: Nutzung von Mikrobiom-Genomdaten zur Erforschung neuer Arzneimittelziele und natürlicher Produkte, Beschleunigung Arzneimittelentwicklung und Entdeckungsbemühungen.
- Biosecurity und Kontrolle: Wachsamkeit bei der Überwachung und präventive Erkennung potenzieller Bioterrorismus- oder Biosecurity-Bedrohungen zur Gewährleistung der öffentlichen und nationalen Sicherheit.
- Lebensmittelsicherheit Überwachung: Überwachung und Erkennung von mikrobieller Kontamination in Lebensmitteln, um die Lebensmittelsicherheitsstandards aufrechtzuerhalten und die Gesundheit der Verbraucher zu schützen.
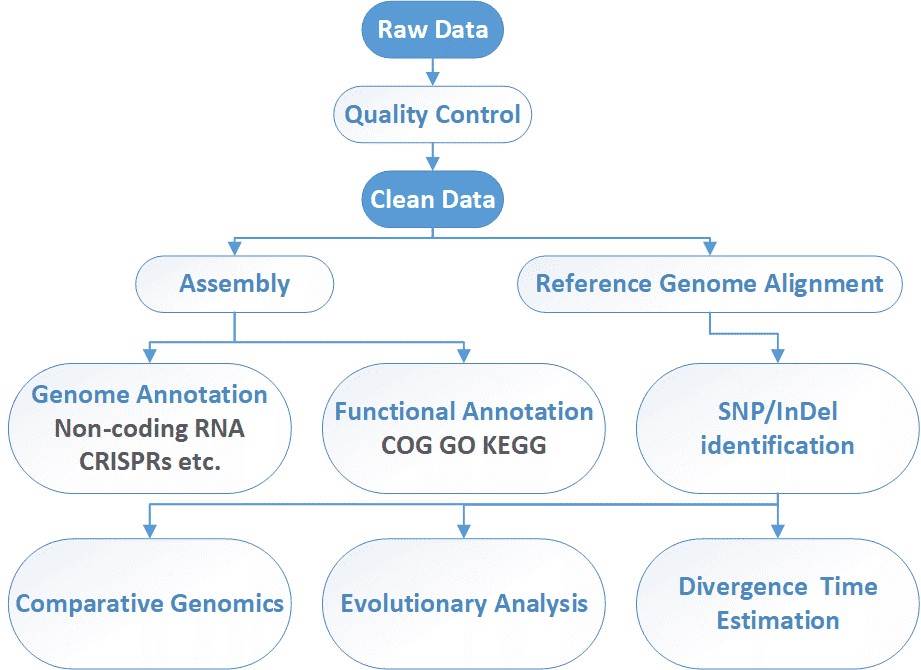
Mikrobielle Ganzgenom-Sequenzierungs-Workflow
CD Genomics kann integrierte Genomsequenzierungsdienste für Bakterien, Hefen, Pilze, Phagen und Viren anbieten. Unser hochqualifiziertes Expertenteam führt ein hochwertiges Management nach jedem Verfahren durch, um zuverlässige und unvoreingenommene Ergebnisse mit dem Illumina HiSeq und/oder dem PacBio SMRT-System zu gewährleisten. Der allgemeine Arbeitsablauf für die gesamte Mikrobengenomsequenzierung ist unten skizziert.

Dienstspezifikation
Musteranforderungen
|
|
 |
Sequenzierung
|
 |
Datenanalyse
Wir bieten maßgeschneiderte bioinformatische Analysen an:
|
Analyse-Pipeline
Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details zur mikrobiellen Gesamtengenomsequenzierung für Ihr Schreiben (Anpassung)
Referenz:
- Fraser C. M., u. a.Mikrobielle Genomsequenzierung. Natur, 2000, 406(6797): 799.
Demo-Ergebnisse
Teilweise Ergebnisse sind unten aufgeführt:
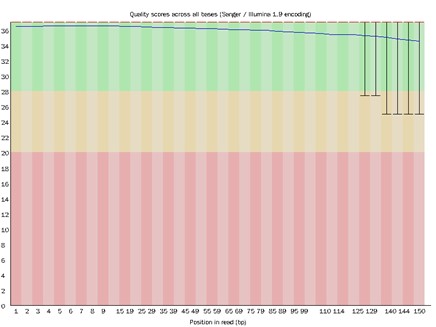
Verteilung der Basisqualität.
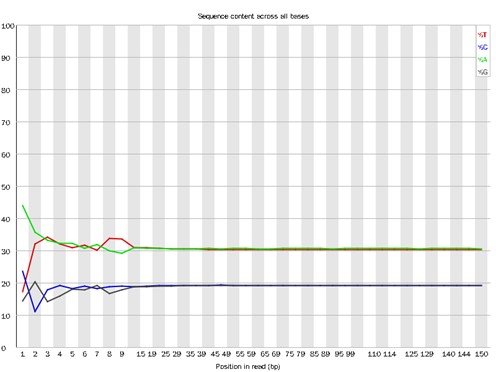
Verteilung von Basisinhalten.
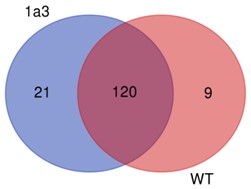
Geteilte SNP-Nummer zwischen Proben.
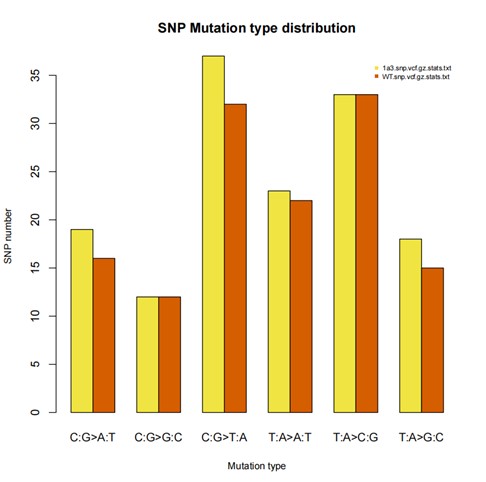
Verteilung der SNP-Mutationsarten.
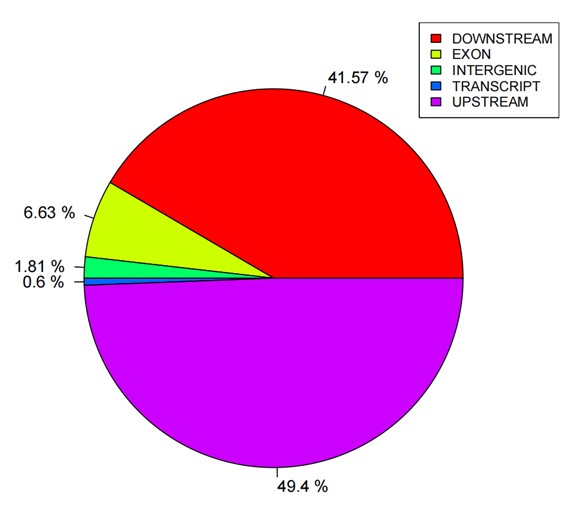
Statistik-Kreisdiagramm der SNP-Anmerkungen.
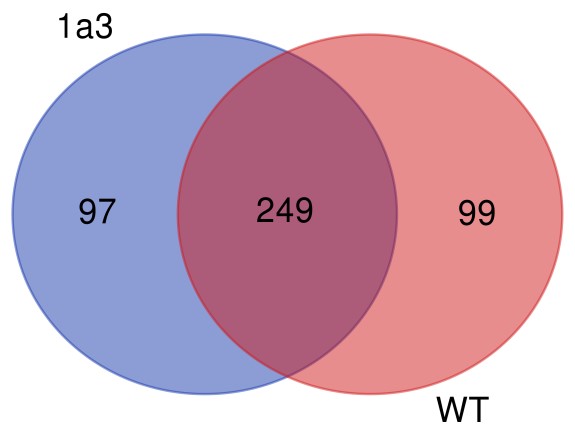
Geteilte InDel-Zahl zwischen Proben.
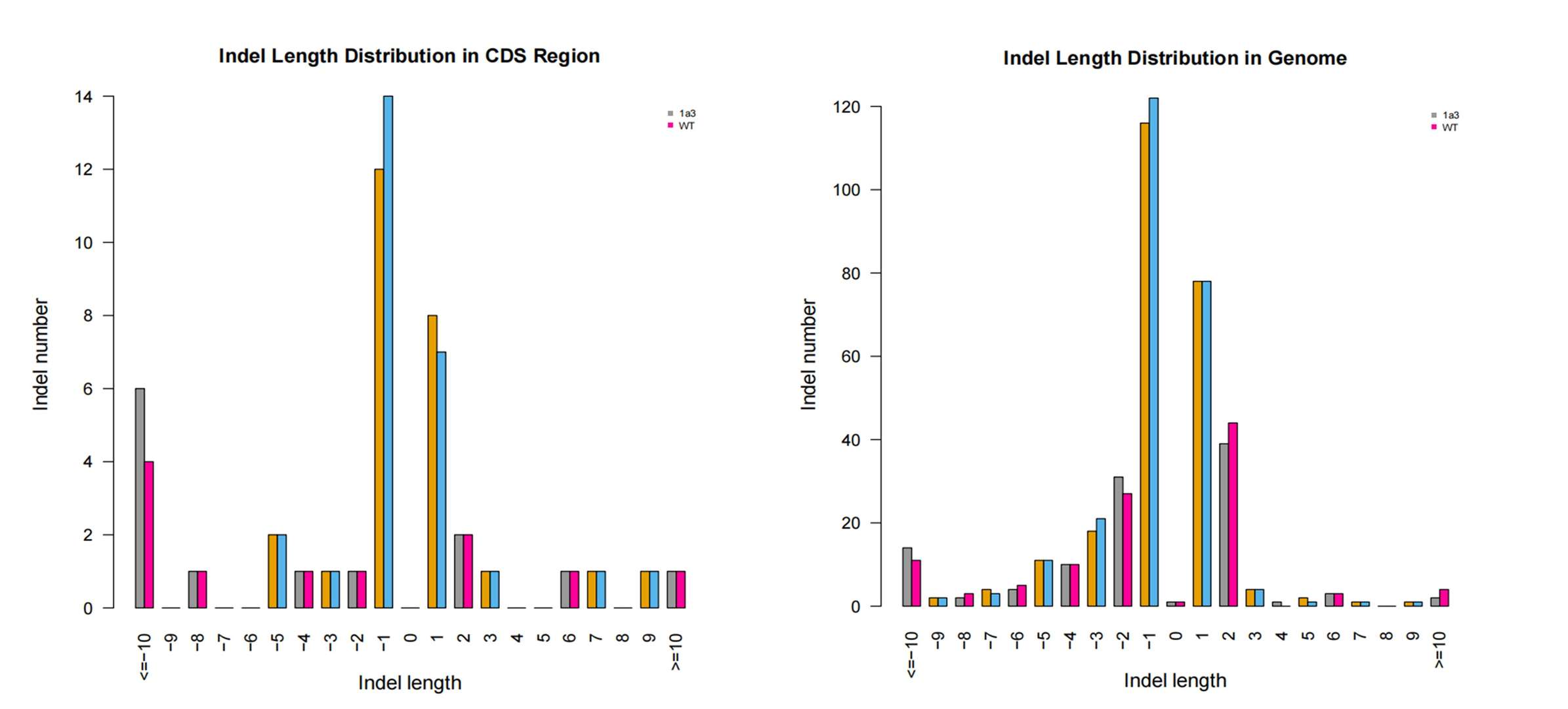
InDel-Längenverteilung sowohl im gesamten Genom als auch in den CDS-Regionen.
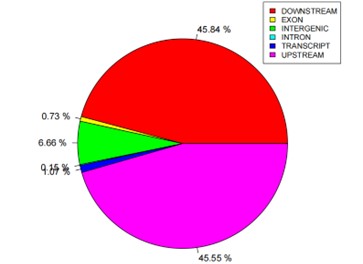
Statistikspie von InDel-Anmerkungen.
FAQs zur mikrobiellen Gesamten Genomsequenzierung
1. Wie bereitet man Proben vor?
Sie können reine kultivierte Mikroorganismen oder extrahierte genomische DNA-Proben für die mikrobielle Ganzgenomsequenzierung einreichen. Für kultivierte Mikroorganismen sollten die gereinigten Zellen in einem 1,5 mL-Röhrchen ohne Medium zentrifugiert werden. Wir benötigen mindestens 1 Million Zellen, und je mehr Zellen Sie anbieten können, desto besser. Für genomische DNA-Proben wird eine empfohlene Menge von 2 µg oder mehr mit einer Konzentration von mehr als 20 ng/µl empfohlen. Das Verhältnis von OD260/OD280 liegt zwischen 1,8 und 2,0. Proben sollten gefroren auf Trockeneis versendet werden.
Stuhlproben: Lagern Sie die Stuhlproben unter -80℃. Empirisch tragen frische Stuhlproben zur Isolierung bevorzugter DNA bei.
2. Wie kann die Genauigkeit der Genomassemblierung bei der Nutzung des PacBio SMRT-Systems erhöht werden?
Die Rohlesefehlerquote von PacBio SMRT System liegt mit etwa 14% erheblich höher im Vergleich zur Fehlerquote von 0,1 bis 1% anderer führender Sequenzierungssysteme. Das Fehlermodell ist jedoch stochastisch, sodass in der Konsenssequenz sehr hochwertige Reads über alle Basen hinweg erzielt werden können. Darüber hinaus ist die SMRT-Sequenzierung Das System ist in der Lage, Regionen mit hohem GC-Gehalt zu sequenzieren, was zu einer viel gleichmäßigeren Abdeckung des Genoms führt.
Es gibt drei Möglichkeiten, die Genauigkeit der Genomassemblierung sicherzustellen: (i) vor der Assemblierung die Sequenzen in der Konsenssequenz korrigieren; (ii) die Ergebnisse der Sequenzassemblierung unter Verwendung von Sequenzierungsdaten korrigieren; (iii) die Ergebnisse der Sequenzassemblierung unter Verwendung von hochqualitativen Daten korrigieren. Next-Generation-Sequenzierung Daten. Nach den drei Korrekturen kann die Genauigkeit der endgültigen Sequenzassemblierung 99,99 % erreichen.
3. Wie erreicht man eine Null-Lücke?
Derzeit kann die vollständige Sequenzkarte von mehr als 90 % der Bakterienstämme durch die Kombination von Illumina HiSeq und erstellt werden. PacBio SMRT Systemen. Die vollständige Sequenzkarte der restlichen 10 % der Bakterienstämme kann mit Sanger-Sequenzierung Daten.
4. Kann eine vollständige Genomassemblierung auch in Regionen mit hohem oder niedrigem GC-Gehalt sowie in repetitiven Sequenzen erreicht werden?
Das Pacbio RS II-System kann die oben genannten Herausforderungen überwinden. CD Genomics hat hunderte von Fällen der Bakteriengenomassemblierung ohne Lücken abgeschlossen.
5. Was sind die Vorteile der mikrobiellen Gesamtensequenzierung des Genoms in der klinischen Praxis?
Whole-Genome-Sequenzierung ist ein routinemäßiges Werkzeug für die klinische Mikrobiologie. Es kann die Diagnosetätigkeit verkürzen und die Kontrolle sowie Behandlung verbessern. Es beschreibt und verbessert unser Verständnis von mikrobieller Evolution, Ausbrüchen und Übertragungsereignissen. Die herkömmlichen Verfahren für Whole-Genome-Sequenzierung beinhaltet oft mehrere Anbau- und Inkubationsschritte, gefolgt von der Identifizierung der Arten, Empfindlichkeitstests und Typisierung, was mehrere Wochen dauern kann. Eine Reihe anderer Ansätze (wie PCR-basierte Methoden) sind kostengünstig und schnell, aber in der Sensitivität begrenzt. Obwohl die Ganzgenomsequenzierung immer noch zu teuer ist, werden die Preise und die Bearbeitungszeiten höchstwahrscheinlich aufgrund des Wettbewerbs unter den Sequenzierungsplattformen sinken.
Referenz:
- Hasman H., u. a.Schnelle Ganzgenomsequenzierung zur Detektion und Charakterisierung von Mikroorganismen direkt aus klinischen Proben. Journal für klinische Mikrobiologie, 2013: JCM. 02452-13.
Fallstudien zur mikrobiellen Gesamtengenomsequenzierung
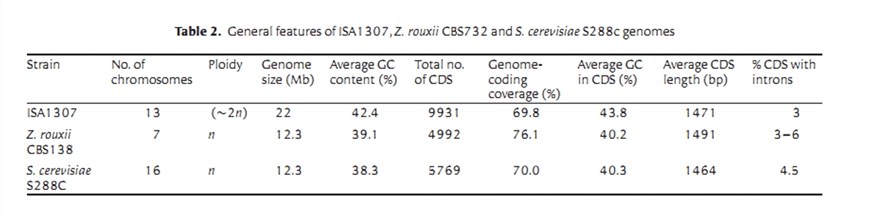
Die Genomsequenz des hochsäurebeständigen interspezifischen Hybridstamms ISA1307, abgeleitet von Zygosaccharomyces bailii, isoliert aus einer Sektanlage.
Zeitschrift: DNA-Forschung
Impactfaktor: 5,404
Veröffentlicht: Juni 2014
Hintergrund
Diese Arbeit beschrieb die Genomsequenzierung und Annotation des Hefestamms ISA1307, der aus einem Sektproduktionsbetrieb isoliert wurde. Dieser Stamm, der früher als der Zygosaccharomyces bailii Art, ist ein interspezifischer Hybrid zwischen Z. bailii und eine eng verwandte Art.
Methoden
- Die prototrophen Hefestämme ISA1307
- Z. bailii ATCC58445T
- S. cerevisiae BY4714 und BY4743
- Quantifizierung von genomischer DNA
- Pulsfeld-Gelelektrophorese (PFGE)
- Whole-Genome-Shotgun-Sequenzierung
- Illumina
- Karyotypisierung des ISA1307-Stammes
- Genomassemblierung und Annotationen
Ergebnisse
1. Der ISA1307 Hybridstamm
Der Vergleich der Sequenzen der Hauskeeping-Gene (einschließlich RPB1, RPB2, EF1-α und β-Tubulin) ergab, dass der Stamm ISA1307 ein interspezifischer Hybrid zwischen Z. bailii und einer eng verwandten Art ist.
2. Genomassemblierung
Die Quantifizierung durch Durchflusszytometrie ergab, dass die geschätzte Größe von ISA1307 etwa 22,0 Mb beträgt, wobei S. cerevisiae BY4741 und BY4743 als Kalibrierungskurve verwendet wurden (Abbildung 1A). Es wurde ein PFGE-Profiling von ISA1307 durchgeführt, und es wurden 13 chromosomale Bänder beobachtet, die Größen von 733 bis 2120 Mb aufwiesen (Abbildung 1B).
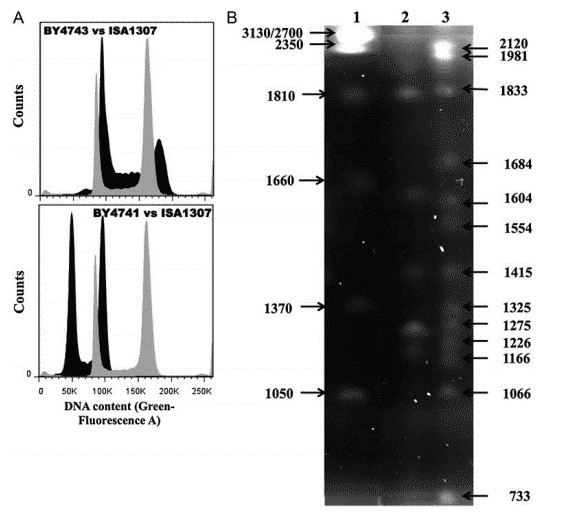 Abbildung 1. Schätzung der Genomgröße und Karyotypisierung des ISA1307-Stamms. (A) Histogramm der repräsentativen Zellanalyse. (B) Karyotyp des Referenzstamms Z. bailii ATCC58445 (Spalte 2) und des ISA1307-Stamms (Spalte 1).
Abbildung 1. Schätzung der Genomgröße und Karyotypisierung des ISA1307-Stamms. (A) Histogramm der repräsentativen Zellanalyse. (B) Karyotyp des Referenzstamms Z. bailii ATCC58445 (Spalte 2) und des ISA1307-Stamms (Spalte 1).
Das endgültig rekonstruierte Genom des ISA1307-Stamms ist auf 154 Scaffold verteilt. Die Summe aller Scaffold-Größen beträgt 21.241.152 bp, was 96 % der Genomgröße entspricht, die durch Flusszytometrie geschätzt wurde (Tabelle 1).

3. Anmerkung
Insgesamt werden 9.925 Gene vorhergesagt, die vom Stamm ISA1307 kodiert werden, darunter 4.385 duplizierte Gene und 1.155 Gene mit einfacher Kopie. Die vorhergesagten Funktionen betreffen "Stoffwechsel und Energieerzeugung", "Protein-Faltung, -Modifikation und -Zielgerichtung" sowie "Biogenese zellulärer Komponenten". Die Autoren untersuchten weiter die Gene und Proteine des ISA1307, die an den oben genannten Funktionen beteiligt sind.
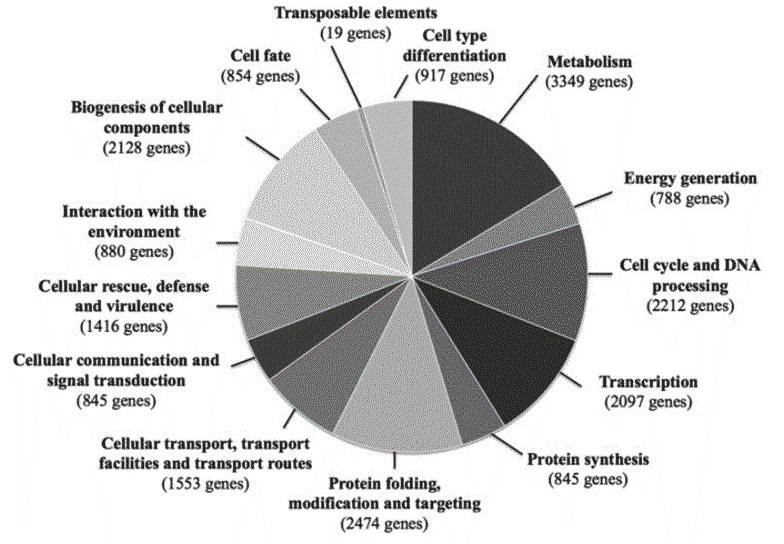 Abbildung 2. Funktionale Klassen von Genen, die voraussichtlich im Genom des ISA1307-Stammes kodiert sind.
Abbildung 2. Funktionale Klassen von Genen, die voraussichtlich im Genom des ISA1307-Stammes kodiert sind.
Fazit
Entsprechend wurde ein Kader spezifischer Referenzallele etabliert, der die Sequenzierungstechnologie der dritten Generation nutzt und eine umfassende longitudinale Erkundung von Genloci über mehrere tausend Basenpaare hinweg bietet. Diese neuartige Technik ergänzt die Sequenzierungstechniken der zweiten Generation oder Kurzlesetechniken und erweist sich als entscheidend hilfreich beim Entschlüsseln neuartiger, seltener und null Allele.
Referenz:
- Mira N. P., u. a.Die Genomsequenz des hochsäurebeständigen interspezifischen Hybridstamms ISA1307, abgeleitet von Zygosaccharomyces bailii, der aus einem Sektbetrieb isoliert wurde. DNA-Forschung, 2014, 21(3): 299-313.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Assoziation des Prophagen BTP1 und des durch den Prophagen kodierten Gens bstA mit der Anti-Virulenz von Salmonella Typhimurium ST313
Journal: Pflanzenkrankheiten
Jahr: 2020
Breitbandige, potente und langlebige Ceria-Nanopartikel inaktivieren die Infektiosität von RNA-Viren, indem sie die Virion-Oberflächen anvisieren und die Virus-Rezeptor-Interaktionen stören.
Journal: Moleküle
Jahr: 2023
Überexpression von UV-Schaden-DNA-Reparaturgenen und Persistenz von Ribonukleinsäure tragen zur Widerstandsfähigkeit getrockneter Biofilme des Wüsten-Cyanobakteriums Chroococcidiopsis bei, die einem marsähnlichen UV-Fluss und langfristiger Austrocknung ausgesetzt sind.
Zeitschrift: Front. Mikrobiol.
Jahr: 2019
Die Gene für die Biosynthese und den Export von Putrescin sind entscheidend für das normale Wachstum von avian pathogenen Escherichia coli.
Journal: BMC Mikrobiologie
Jahr: 2018
Phänotypische und Entwurf-Genomsequenzanalysen eines Paenibacillus sp., isoliert aus dem Magen-Darm-Trakt eines nordamerikanischen Grauwolfs (Canis lupus)
Journal: Angewandte Mikrobiologie
Jahr: 2023
Klassifikation der Gattungen der Pasteurellaceae anhand konservierter vorhergesagter Proteinsequenzen
Internationales Journal für systematische und evolutionäre Mikrobiologie
Jahr: 2018
Mehr ansehen Artikel, die von unseren Kunden veröffentlicht wurden.


