
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Whole-Genome-SNP-Genotypisierung
SNP, eine Abkürzung für Single Nucleotide Polymorphism (einzelner Nukleotid-Polymorphismus), bezeichnet eine Form des genetischen Polymorphismus, die aus einer Mutation in der DNA-Sequenz des Genoms resultiert, ausgelöst durch eine einzelne Nukleotidveränderung, die den Austausch, die Transversion, die Insertion oder die Deletion einer einzelnen Base umfassen kann. Grundsätzlich können SNPs als bi-allelisch, tri-allelisch oder tetra-allelisch auftreten, jedoch sind die letzteren beiden in der praktischen Anwendung äußerst selten und werden als unbedeutend erachtet. Im Allgemeinen liegt der Fokus bei der Bezugnahme auf SNPs auf bi-allelischen Polymorphismen. Diese Variationen können als Transitionen auftreten, exemplifiziert durch die Umwandlung von Cytosin (C) zu Thymin (T) oder dessen komplementärem Strang-Gegenstück, nämlich die Umwandlung von Guanin (G) zu Adenin (A). Alternativ können sie auch als Transversionen auftreten, wie Cytosin zu Adenin (CA), Guanin zu Thymin (GT), Cytosin zu Guanin (CG) oder Adenin zu Thymin (AT). Es ist wichtig zu beachten, dass Transitionen durchweg deutlich höhere Auftretensraten aufweisen als andere Kategorien, wobei SNPs vom Transitionstyp etwa zwei Drittel des Gesamtanteils ausmachen.
Warum SNP-Genotypisierung erkennen?
Häufige SNPs haben das Potenzial, sowohl in kodierenden als auch in nicht kodierenden Regionen des Genoms aufzutreten. Während ihr Vorkommen in kodierenden Regionen relativ selten ist, macht ihre Fähigkeit, die Funktionalität von Genen zu beeinflussen und somit Veränderungen in biologischen Merkmalen zu bewirken, sie von größter Bedeutung im Bereich der genetischen Krankheitsforschung. Als genetische Marker der dritten Generation sind SNPs gleichmäßig über die gesamten genomischen Landschaften von Tieren und Menschen verteilt. Sie weisen eine ausgeprägte Korrelation mit funktionalen Genen auf, haben minimale Mutationsraten und zeigen eine robuste genetische Stabilität. Darüber hinaus eignen sie sich für Hochdurchsatzanalysen und eine automatisierte Durchführung. Es ist bemerkenswert, dass bestimmte SNP-Loci möglicherweise nicht direkt mit der Expression von krankheitsassoziierten Genen korrelieren. Aufgrund ihrer Nähe zu bestimmten pathogenen Genen übernehmen sie jedoch entscheidende Rollen als wichtige genetische Marker in diesen Kontexten.
Mikroarray-basierte SNP-Analyse
Gen-Chips bieten eine Vielzahl von Vorteilen, darunter eine hohe Durchsatzrate, makellose Genauigkeit und benutzerfreundliche Bedienung, was sie besonders geeignet für Anwendungen macht, bei denen die spezifischen Zielorte bereits festgelegt sind. Insbesondere wenn die Anzahl der Zielorte einen bestimmten Schwellenwert überschreitet, zeigen Gen-Chips eine bemerkenswerte Reduzierung der Gesamtkosten bei gleichzeitig gesteigerter Effizienz.
Die Genchip-Technologie basiert auf dem Prinzip der komplementären Basenhybridisierung. Sie umfasst das Entwerfen von Sonden-Sequenzen für bekannte SNP-Loci und das Markieren von DNA-Sonden mit Isotopen oder fluoreszierenden Markern. Diese markierten DNA-Sonden werden mit der Ziel-DNA hybridisiert, um biologische Informationen zu erhalten. Genchips nutzen die Mikroarray-Technologie, um Millionen von Sonden auf einem Siliziumchip oder einer Nylonmembran in der Größe eines Mikroskopträgers zu integrieren. Dies ermöglicht die gleichzeitige Analyse von 5.000 bis 1.000.000 Genotypen für eine Person.
Unser Mikroarray-basierte SNP-Genotypisierungsdienste bestehen aus Affymetrix SNP-Arrays und Illumina SNP-Arrays. Die Affymetrix Genotypisierungs-Arrays auf dem GeneTitan-Instrument (Axiom-Array-Platten) und dem GeneChip Scanner 3000 7G System (Cartridge-Arrays) können für eine Vielzahl von Anwendungen verwendet werden – für wenige und viele Proben von gezielten bis hin zu genomweiten Anwendungen durch die Auswahl spezifischer, vorgefertigter Genotypisierungs-Arrays. Die hochdichte Genotypisierung auf Illumina Infinium oder GoldenGate®-Arrays ermöglicht leistungsstarke genomweite Assoziationsstudien (GWAS) und kann Punktmutationen und Kopienzahlvarianten genau erkennen. Derzeit bietet Affymetrix Genotypisierungs-Arrays für Nutztiere und Aquakulturspezies (Büffel, Rinder, Hühner, Schweine, Lachs und Forellen), Pflanzen (Baumwolle, Mais, Sojabohnen, Erdbeeren und Weizen) sowie biomedizinische Modellorganismen (Mensch, Hund, Maus und Arabidopsis thaliana) an, während Illumina Genotypisierungs-BeadArrays für menschliche und nicht-menschliche Spezies (Rinder, Hunde, Mais, Schweine und Schafe) vermarktet. SNP-Arrays wurden in einigen Fällen als bevorzugte Technik angesehen, aufgrund ihrer hohen Dichte, Assaygenauigkeit, einfachen Datenanalyse und einfachen Datenübertragung zwischen Forschungsprogrammen. Diese kommerziell verfügbaren SNP-Arrays oder -Chips können jedoch nicht leicht modifiziert werden, um individuellen experimentellen Designs zu entsprechen. Darüber hinaus können relevante Forschungen für Spezies, die keine kommerziell verfügbaren SNP-Arrays/Chips haben, nicht durchgeführt werden.
Die technischen Grundlagen von Gen-Chips bieten die folgenden Vorteile:
Erhöhter Durchsatz: Gen-Chips ermöglichen die gleichzeitige Untersuchung einer umfangreichen Reihe von genetischen Loci, mit Kapazitäten von bis zu 1.000.000 Loci.
Strenge Standardisierung und Reproduzierbarkeit: Jeder experimentelle Durchlauf liefert präzise genetische Informationen an vordefinierten Positionen, beseitigt inhärente Zufälligkeit und gewährleistet konsistente Ergebnisse.
Beispielhafte Genauigkeit: Der Chipproduktionsprozess ist von strengen Qualitätsbewertungen geprägt, wobei jeder Erkennungsort wiederholten Prüfungen unterzogen wird, was die Ergebnisse von bemerkenswerter Präzision untermauert.
Effizienz: Experimente mit Gen-Chips werden beschleunigt, wobei Ergebnisse in nur 2 Tagen erreichbar sind, was zur Schnelligkeit der Forschung beiträgt.
Kosten-Effizienz: Gen-Chips bieten eine kosteneffiziente Lösung, die besonders für großangelegte, hochdurchsatzfähige genetische Profilierungsprojekte geeignet ist.
Anpassungsfähigkeit: Genchips können entsprechend spezifischer Zielorte angepasst werden, wobei Mikroarray-Chips im Vergleich zu in situ synthetisierten Gegenstücken eine verbesserte Flexibilität bei der Anpassung bieten.
NGS-basierte SNP-Genotypisierung
NGS (Next-Generation Sequencing) Technologien sind leistungsstarke Werkzeuge für SNP-Genotypisierung, da sie effizient und genau Tausende von SNPs entdecken und genotypisieren können, um quantitative, funktionale und evolutionäre Genomik bei Menschen, Tieren und Pflanzen zu untersuchen.
NGS ist eine DNA-Sequenzierungstechnologie, die sich aus PCR und Genchips entwickelt hat. Im Vergleich zur Sanger-Sequenzierung ermöglicht die NGS-Sequenzierung eine reversible Beendigung der Enden, wodurch Synthese und Sequenzierung gleichzeitig erfolgen können.
Bei NGS müssen einzelne DNA-Moleküle in Cluster amplifiziert werden, die aus identischen DNA-Zusammensetzungen bestehen. Diese Cluster werden dann synchron repliziert, um die Fluoreszenzsignalintensität zu erhöhen, was das Lesen der DNA-Sequenz erleichtert. Mit zunehmender Leselänge nimmt die Kohäsion der Clusterreplikation ab, was zu einem Rückgang der Sequenzierungsqualität führt. Daher hat NGS typischerweise kürzere Leselängen, die in der Regel 500 Basenpaare nicht überschreiten. Nach der Sequenzierung müssen die Informationen aus fragmentierten DNA-Segmenten zusammengefügt werden, und der Sequenzierungsprozess hat eine Fehlerquote im Bereich von 0,1 % bis 15 %, was die Genauigkeit verringert.
Die verkürzten Leselängen in der NGS ermöglichen die gleichzeitige Bewertung von Hunderttausenden bis hin zu Millionen von Gen-Sequenzen innerhalb eines einzigen Sequenzierungslaufs. Dieser Durchbruch überwindet effektiv die mit der niedrigen Durchsatzrate verbundenen Einschränkungen, die für Sequenzierungsmethoden der ersten Generation charakteristisch waren. Ein einzelner NGS-Lauf kann effizient Multi-Locus-Genotypisierungs-Experimente durchführen, insbesondere bei umfangreichen Stichproben, und bietet dabei erhebliche Zeitersparnisse und Kostenreduzierungen. Folglich hat diese Technologie eine breite Akzeptanz im Bereich der Hochdurchsatz-Gen-Sequenzierung gefunden.
Es gibt verschiedene Methoden zur Genotypisierung von Einzelne Nukleotid-Polymorphismen (SNP) basierend auf der Next-Generation-Sequenzierung.
Whole-Exom-Sequenzierung (WES): WES ist ein Verfahren, das NGS verwendet, um nur die exonspezifischen Regionen des menschlichen Genoms zu sequenzieren, die Proteine kodieren. Während es hauptsächlich zur Untersuchung der Beziehung zwischen Genmutationen und Krankheiten eingesetzt wird, kann es auch zur SNP-Genotypisierung verwendet werden, da Exons viele SNP-Loci enthalten.
Whole-Genome-Sequenzierung (WGS): WGS umfasst die Sequenzierung aller Regionen des gesamten Genoms, einschließlich kodierender und nicht kodierender Regionen. Diese Methode kann verwendet werden, um alle SNPs im Genom zu erkennen und zu typisieren, und bietet umfassende genetische Informationen.
Gezielte SequenzierungDer gezielte Sequenzierungsansatz umfasst die spezifische Sequenzierung von Genen oder SNP-Loci von besonderem Interesse, wodurch der Erwerb von hoch fokussierten SNP-Daten ermöglicht wird. Diese Methodik kann so angepasst werden, dass sie die selektive Zielverfolgung von SNPs umfasst, die mit bestimmten Krankheiten assoziiert sind, was eine sorgfältige Untersuchung genetischer Variationen ermöglicht, die für die untersuchte Pathologie relevant sind.
RNA-Sequenzierung (RNA-Seq)RNA-Seq, das hauptsächlich zur Untersuchung von Genexpressionsprofilen genutzt wird, bietet eine vielseitige Anwendung, die die Erkennung von Einzelne Nukleotid-Polymorphismen (SNPs) umfasst. Dieser facettenreiche Ansatz, der auf der Analyse von Gen-Transkripten basiert, ermöglicht die Erkennung von SNP-Expression und Variationen auf RNA-Ebene und bietet somit ein umfassendes Verständnis der genetischen Vielfalt und Dynamik in diesem Kontext.
MultiplexingNGS-Technologien ermöglichen häufig das gleichzeitige Sequenzieren mehrerer Proben, was zu einer erhöhten Effizienz und Kostenreduktion führt. Diese Methodik erweist sich als äußerst vorteilhaft im Kontext großangelegter Genotypisierungsbemühungen von einzelnen Nukleotidpolymorphismen (SNP), einschließlich genomweiter Assoziationsstudien (GWAS).
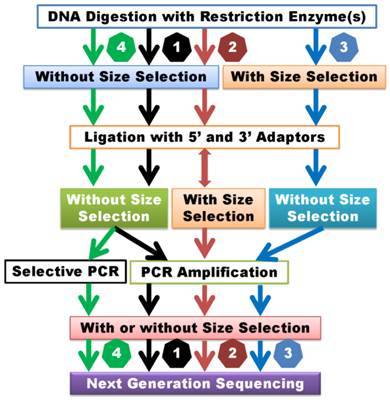
Genotypisierung durch Sequenzierung (GBS): GBS ist ein Hochdurchsatzansatz, der auf der Sequenzierungstechnologie der zweiten Generation basiert. Diese Methodik ermöglicht die gleichzeitige Sequenzierung von DNA-Proben mehrerer Individuen und die Bestimmung ihrer jeweiligen Genotypen. Das GBS-Protokoll umfasst die Bibliotheksvorbereitung, die gezielte Sequenzierung spezifischer genomischer Regionen, die durch die DNA-Digestion mit Restriktionsenzymen erreicht wird, und die Verwendung von Hochdurchsatz-Sequenzierern zur Erstellung umfangreicher Datensätze, die reich an Informationen über einzelne Nukleotidpolymorphismen (SNP) sind.
dd-RAD (Double Digest Restriction-site Associated DNA Sequenzierung)dd-RAD stellt eine zusätzliche SNP-Genotypisierungstechnik dar, die auf der Sequenzierungstechnologie der zweiten Generation basiert. Dieses Verfahren umfasst die Verwendung von zwei unterschiedlichen Restriktionsenzymen, um DNA zu spalten, gefolgt von der Sequenzierung der gespaltenen Produkte zur Erkennung und Genotypisierung von SNP-Loci.
2b-RAD2b-RAD ist eine Methode zur Sequenzierung von reduzierten Genomdarstellungen oder eine mit Restriktionsstellen assoziierte DNA-Sequenzierungsstrategie, um genetische Varianten kosteneffektiv zu entdecken und zu genotypisieren, die sowohl für Modellorganismen mit bekannten Genomen als auch für Nicht-Modellorganismen mit unbekannten Genomen anwendbar sind. Diese Technik wird definiert als die Bibliothekskonstruktion durch Verdau von DNA mit Restriktionsenzymen und die anschließende Analyse der Bibliothek mit Illumina-Sequenzierungsplattformen.
PCR-basierte SNP-Genotypisierungstechniken
PCR bietet hohe Sensitivität, niedrige Kosten für Szenarien mit einer begrenzten Anzahl von zu testenden Loci und kürzere Erkennungszeiten, die typischerweise innerhalb von 2-4 Stunden abgeschlossen sind. Es ist operationell unkompliziert. Allerdings hat es Einschränkungen, wie die Erkennung eines einzelnen Locus, niedrige Durchsatzraten (normalerweise zwischen wenigen und einigen hundert Loci) und die Unfähigkeit, andere Mutationen zu erkennen, die möglicherweise in der DNA vorhanden sind. Falsch-negative und falsch-positive Ergebnisse sind möglich, was es für Szenarien mit einer kleinen Anzahl von zu testenden Loci und geringeren Durchsatzanforderungen geeignet macht.
Ligase-Detektionsreaktion (LDR) GenotypisierungDieses Verfahren verwendet hauptsächlich die Ligase-Detektionsreaktion (LDR) Technologie. Taq-DNA-Ligase wird eingesetzt, um die Reaktion zu erleichtern, die nur stattfindet, wenn zwei kurze Nukleotid-Sonden, eine von jedem Strang, vollständig komplementär zur Ziel-DNA-Sequenz sind, ohne Lücken. Die Detektion erfolgt durch fluoreszierendes Scannen der Fragmentlängen, was die Identifizierung von SNP-Loci ermöglicht.
Multiplex SNaPshot SNP-GenotypisierungSNaPshot SNP-Genotypisierung ist ein Verfahren, das gleichzeitig mehrere PCRs verwendet, um die genetische Typisierung mehrerer bekannter SNP-Loci durchzuführen. Es nutzt DNA-Sequenzierungsenzyme, vier fluoreszenzmarkierte ddNTPs, Verlängerungsprimer unterschiedlicher Längen, die an der polymorphen Stelle angrenzen, und PCR-Produktvorlagen. Die Primerverlängerung endet nach einer einzelnen Base, und nach der Gelelektrophorese auf einem ABI 3730-Sequenzer zeigen die Farben der Peaks die inkorporierte Base an, wodurch der Genotyp der Probe und der SNP-Locus bestimmt werden, der dem Verlängerungsprodukt basierend auf der Peak-Mobilität entspricht.
Taqman-ProbenmethodeDieses Verfahren beinhaltet das Hinzufügen von zwei Sonden mit unterschiedlichen fluoreszierenden Markierungen zum PCR-Reaktionssystem. Jede Sonde ergänzt vollständig eines der beiden Allele. Normalerweise wird aufgrund der engen Nähe der fluoreszierenden Gruppe am 5'-Ende und der Quencher-Gruppe am 3'-Ende der Sonden die Fluoreszenz gequencht. Während die PCR voranschreitet, wird die vollständig komplementäre Sonde zum Template allmählich durch die 5'→3' Exonukleaseaktivität der Taq-DNA-Polymerase gespalten, was zur Trennung der fluoreszierenden Gruppe am 5'-Ende von der Quencher-Gruppe am 3'-Ende führt und die Fluoreszenz dequencht. Im Gegensatz dazu wird die Sonde, die das andere Allel repräsentiert und nicht perfekt mit dem Template übereinstimmt, nicht effizient gespalten, und es wird keine Fluoreszenz detektiert. Die Erkennung des SNP-Lokus erfolgt durch die Messung von Änderungen der Fluoreszenzwerte mit der entsprechenden Instrumentierung.
PCR-ARMS (Amplification Refractory Mutation System): ARMS ist ein PCR-Amplifikationsverfahren, das für spezifische SNPs entwickelt wurde. Durch das Design der Primer amplifiziert es selektiv DNA-Fragmente, die den spezifischen SNP enthalten oder nicht enthalten.
PCR-basierte Techniken bieten eine Vielzahl von Optionen für die SNP-Genotypisierung, die sich durch ihre einzigartigen Merkmale und Anpassungsfähigkeit an spezifische Forschungskontexte unterscheiden.
Massen spektrometriebasierte SNP-Genotypisierung
MassARRAY SNP-Genotypisierung: Die Matrix-unterstützte Laserdesorption/Ionisation Zeit-of-Flight Massenspektrometrie (MALDI-TOF MS) dient als grundlegende Technologie für dieses Verfahren. Zunächst wird PCR eingesetzt, um Genabschnitte mit SNPs zu amplifizieren. Anschließend erfolgt eine Einzelbasenerweiterung unter Verwendung von sequenzspezifischen Primern. Danach wird das Probenanalyte mit der Chipmatrix co-kristallisiert und durch einen Nanosekundenlaserimpuls (10-9s) innerhalb eines Vakuumrohrs angeregt. Dieser Prozess führt zur Desorption von Nukleinsäuremolekülen, wodurch sie in einfach positiv geladene Ionen umgewandelt werden. Aufgrund der umgekehrten Beziehung zwischen der Ionmasse und der Flugzeit des Ions in einem elektrischen Feld wird das genaue Molekulargewicht des Probenanalyten ermittelt, indem die Flugzeit der Nukleinsäuremoleküle im Vakuumrohr gemessen wird. Dadurch können SNP-Stelleninformationen zuverlässig detektiert werden. Dieses Verfahren eignet sich gut für die mittel- bis hochdurchsatz SNP-Analyse, insbesondere bei mehr als zehn SNP-Stellen.
CD Genomics verpflichtet sich, SNP-Genotypisierung auf genomweiter Ebene und auf erschwingliche Weise anzubieten. Unsere erfahrenen Wissenschaftler können professionelle Unterstützung für Ihr Projekt bieten, um Ihre Forschungsziele und Ihr Budget zu erfüllen.
Vergleich von SNP-Genotypisierung und RAD-Seq-Methoden
Hier ist eine klare Übersicht der Genotypisierung und RAD-seq-Techniken:
| Methode | genomweit | Fragmentquelle | Größenauswahl | Durchsatz | Bester Anwendungsfall |
|---|---|---|---|---|---|
| RAD-seq (klassisch) | ✅ Ja | Einzelnes Enzym + zufälliges Scheren | Physikalisches Scheren | Mittel | Nicht-Modellspezies ohne Referenzgenom |
| ddRAD-seq | ✅ Ja | Zwei Enzyme + größenselektierte Fragmente | Gel-basiert, präziser Bereich | Hoch | Ausgewogene SNP-Entdeckung mit einheitlicher Abdeckung |
| 2b-RAD | ✅ Ja | Typ IIB-Enzym → einheitliche 33–36 bp-Tags | Keine Größenwahl erforderlich | Sehr hoch | Hochdichte SNP-Kartierung, selbst auf degradiertem DNA |
| PCR-LDR-Genotypisierung | ❌ Nur gezielt | PCR + Ligase-Nachweis | N/V | Niedrig | Validierung bekannter SNPs für ein oder wenige Loci |
| MassARRAY Genotypisierung | ❌ Nur gezielt | PCR + Einzelbasenerweiterung + MALDI-TOF-MS | PCR-basierte Primer | Mittel–Hoch | Multiplexvalidierung von ~10–100 SNPs |
Wählen Sie eine Methode basierend auf:
- 2b-RAD oder ddRAD-seq für die genomweite Entdeckung
- Klassisches RAD-seq für nicht-referenzierte Arten
- PCR-LDR für kleinmaßstäbige, hochpräzise SNP-Tests
- MassARRAY für mittelständische, multiplexierte Validierung


