
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Epigenomik-Sequenzierung (DNA/RNA-Methylierungsanalyse)
CD Genomics bietet Plattformen für die genomweite Epigenom-Analyse an, die jeweils so konzipiert sind, dass sie eine Vielzahl von Probenarten aufnehmen und Ihren spezifischen Forschungsbedürfnissen entsprechen, sodass Forscher epigenetische Veränderungen leicht untersuchen können. Diese Informationen werden uns nicht nur helfen, die Rolle der DNA-Methylierung zu verstehen, sondern auch Ziele für therapeutische Behandlungen zu identifizieren.
Was sind epigenetische Modifikationen?
Epigenetische Modifikationen sind reversible Modifikationen, die die Genexpression beeinflussen, ohne die DNA-Sequenz zu verändern, und können während der Zellteilung vererbt werden. Zwei der am besten charakterisierten epigenetischen Modifikationen sind die DNA-Methylierung und die Chromatinmodifikation. Epigenetische Modifikationen spielen eine wichtige Rolle bei der Genexpression und -regulation und sind an zahlreichen zellulären Prozessen wie Differenzierung, Entwicklung und Tumorigenese beteiligt. DNA-Methylierung wird am häufigsten an der C5-Position von Cytosin beobachtet, gefolgt von Guanin (CpG-Stelle) bei Wirbeltieren oder an Nicht-CpG-Stellen wie CHG und CHH in Pflanzen oder embryonalen Stammzellen von Säugetieren. Die DNA-Methylierung wird von DNA-Methyltransferasen (DNMT1, DNMT3a und DNMT3b) etabliert und aufrechterhalten.
Die Epigenomik kann in zwei Hauptkategorien unterteilt werden:
1. EpigenomDas Epigenom umfasst eine Vielzahl chemischer Modifikationen, die auf DNA und Histonen innerhalb der Chromatinstruktur auftreten, zusätzlich zu strukturellen Veränderungen im Chromatin selbst. Diese Modifikationen, unabhängig von der zugrunde liegenden genomischen Sequenz, liefern wichtige regulatorische Informationen. Die folgenden sind zentrale Komponenten des Epigenoms:
(1) DNA-ModifikationenBeinhaltet 5-Methylcytosin (5mC) und 5-Hydroxymethylcytosin (5hmC).
(2) HistonmodifikationenUmfasst Modifikationen wie H3K27me3, H3K4me3 und H3K27ac.
(3) ChromatinveränderungenBeinhaltet Veränderungen, die durch DNA-bindende Proteine wie Transkriptionsfaktoren vermittelt werden.
2. EpitranskriptomDieser Begriff bezieht sich allgemein auf alle posttranskriptionalen Modifikationen, die die RNA-Sequenz selbst nicht verändern. Derzeit wurden über 100 verschiedene chemische Modifikationen auf RNA identifiziert, darunter N6-Methyladenosin (m6A), N1-Methyladenosin (m1A) und Pseudouridin (Ψ).
DNA-Methylierungs-Sequenzierungstechnologien
DNA-Methylierungsinformationen können während standardmäßiger molekularbiologischer Manipulationen, wie z.B. molekularer Klonierung in Bakterien und PCR, verloren gehen, da die DNA-Methyltransferasen nicht erhalten bleiben. Mehrere Techniken umfassen methylierte DNA-Immunpräzipitation (MeDIP), Bisulfitsequenzierung (BS-seq)und reduzierte Repräsentation Bisulfit-Sequenzierung (RRBS) wurden vorgeschlagen, um DNA-Methylierungsinformationen zu bewahren und gleichzeitig in quantitative und messbare Signale umzuwandeln. Durch die Kombination mit Hochdurchsatz-SequenzierungDiese Techniken haben umfassende und zuverlässige genomweite Informationen über die DNA-Methylierung bereitgestellt. Eine kurze Übersicht und der Arbeitsablauf verschiedener NGS-basierter DNA-Methylierungssequenzierungstechnologien sind in Abbildung 1 dargestellt.
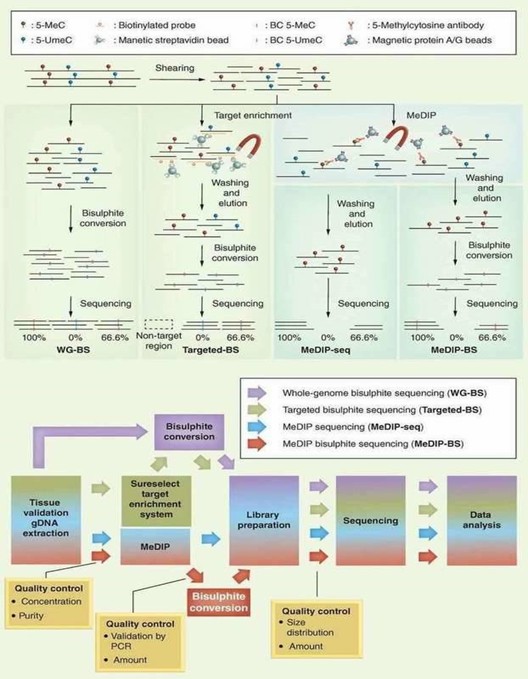 Abbildung 1. Verschiedene auf NGS basierende Methoden zur DNA-Methylierungsanalyse (Jeong u. a.., 2016).
Abbildung 1. Verschiedene auf NGS basierende Methoden zur DNA-Methylierungsanalyse (Jeong u. a.., 2016).
In Bezug auf die Vorzüge und Vorurteile dieser Methoden, BS-Seq und RRBS kann ein DNA-Methylom mit Basisauflösung erzeugen, während MeDIP-seq kann nur relative Anreicherungen spezifischer Regionen im Genom erzeugen. Die Chromatin-Immunpräzipitation (ChIP) bietet ein vorteilhaftes Werkzeug zur Untersuchung der Histonmethylierungsniveaus, die mit einer spezifischen Genpromotorregion zwischen normalen und erkrankten Geweben assoziiert sind. Die Identifizierung der genetischen Ziele von DNA-bindenden Proteinen und die Aufdeckung des Mechanismus der Protein-DNA-Interaktion sind entscheidend für das Verständnis zellulärer Prozesse. Die Chromatin-Immunpräzipitation-Sequenzierung (ChIP-seq) ermöglicht es Ihnen, das Beste aus Ihren Chromatinstudien mit minimaler Sequenzierungsverzerrung herauszuholen.
Wie man RNA-Methylierung nachweist
Sowohl die DNA- als auch die RNA-Methylierung beinhalten die enzymatische Addition einer Methylgruppe (CH3) zu einem spezifischen Atom auf dem DNA- oder RNA-Molekül, katalysiert durch Methyltransferasen. In zellulärer RNA wurden bereits mehr als 100 Arten chemischer Modifikationen identifiziert. Unter diesen gibt es verschiedene Arten der RNA-Methylierung, einschließlich m6A RNA-Methylierung, m5C RNA-Methylierung, m1A RNA-Methylierung und m7G RNA-Methylierung. Derzeit ist die prominenteste und am stärksten angereicherte Art die m6A RNA-Methylierung, die sich auf die Methylierung des Stickstoffatoms an der 6. Position von Adenin in RNA-Molekülen (N6-Methyladenosin, m6A) bezieht. Diese Modifikation ist die häufigste posttranskriptionale Modifikation in eukaryotischer mRNA und macht 80 % der RNA-Methylierungsmodifikationen aus.

Die aktuellen Methoden zur Erkennung von RNA-Methylierung umfassen hauptsächlich Folgendes:
- MeRIP-Sequenzierung (Methylierte RNA-Immunpräzipitation-Sequenzierung)Dieses Verfahren basiert auf dem Prinzip der antikörperspezifischen Bindung an methylierte Basen. Es verwendet m6A-spezifische Antikörper, um methylierte RNA-Fragmente zu immunpräzipitieren und anzureichern, die dann einer Hochdurchsatz-Sequenzierung unterzogen werden, um m6A-Modifikationen zu identifizieren. Allerdings kann dieses Verfahren nur Regionen mit hohen Methylierungsgraden identifizieren und erreicht keine Einzelbasenauflösung für RNA-Methylierung.
- miCLIP (Methylierung Individuelle Nukleotidauflösungs-Kreuzvernetzung und Immunpräzipitation)In dieser Technik wird methyliertes RNA spezifisch an Antikörper gebunden und dann mit ultraviolettem Licht quervernetzt. Die reversen Transkription der quervernetzten RNA führt zu cDNA-Mutationen oder -Trunkierungen, die auf das Vorhandensein von m6A hinweisen. Während miCLIP RNA-Methylierung mit einer Einzelbasenauflösung identifizieren kann, machen die hohen Kosten, die mit der Verwendung von isotopischen Markierungen wie P32 verbunden sind, es weniger praktikabel für den routinemäßigen Laborgebrauch.
- Nanoporen-Sequenzierung TechnologieDiese Sequenzierungstechnologie der dritten Generation identifiziert Basensequenzen basierend auf elektrischen Signalen. Unterschiedlich modifizierte Basen auf RNA verursachen unterschiedliche Grade der Behinderung, während sie durch den Nanoporenkanal passieren, was charakteristische elektrische Signale erzeugt. Durch die Echtzeitüberwachung dieser Signale können die entsprechenden Basentypen und deren Modifikationen bestimmt werden. Somit kann die Nanoporen-Sequenzierung RNA-Methylierung mit einer Einzelbasenauflösung erkennen, ohne dass spezifische Antikörperbindung erforderlich ist.
Unsere Epigenomik-Sequenzierungsdienste
Unsere DNA-Methylierungslösungen umfassen:
Whole-Genome-Bisulfid-Sequenzierung
Die Whole Genome Bisulfite-Sequenzierung bietet einen umfassenden Überblick über die DNA-Methylierungsmuster im gesamten Genom und ermöglicht die Untersuchung von methylierte und unmethylierte Cytosinen.
Gezielte Bisulfit-Sequenzierung
Diese Sequenzierungsmethodik erfasst und untersucht selektiv definierte genomische Regionen und ermöglicht die präzise und kosteneffiziente Analyse von DNA-Methylierungsmustern.
Reduzierte Repräsentation Bisulfit-Sequenzierung
Die reduzierte Repräsentations-Bisulfid-Sequenzierung ist ein Verfahren, das sich auf einen Teil des Genoms konzentriert und ein Gleichgewicht zwischen Genomabdeckung und Kosten-Effektivität für die DNA-Methylierungsanalyse bietet.
MeDIP-Sequenzierung
MeDIP-Sequenzierung erleichtert die Anreicherung und Analyse von methylierten DNA-Fragmenten und bietet wertvolle Einblicke in die Muster der DNA-Methylierung und deren potenzielle funktionale Implikationen.
ChIP-Seq
Die Chromatin-Immunpräzipitation-Sequenzierung (ChIP-Seq) ist eine leistungsstarke Methode zur Identifizierung von Protein-DNA-Interaktionen, zur Aufklärung von Bindungsstellen von Transkriptionsfaktoren und zur Erforschung epigenetischer Modifikationen.
hMeDIP-seq
Hydroxymethylierte DNA-Immunpräzipitations-Sequenzierung (hMeDIP-Seq) wird verwendet, um spezifisch die DNA-Hydroxymethylierung zu untersuchen, ein entscheidendes epigenetisches Merkmal, das an der Genregulation beteiligt ist.
ATAC-Seq
ATAC-Seq liefert Daten zur Chromatinzugänglichkeit und ermöglicht die Identifizierung sowohl offener als auch geschlossener Chromatinregionen im Genom.
NGS-BSP
Next-Generation Sequencing von bisulfit-konvertierter DNA (NGS-BSP) ist ein Verfahren zur umfassenden Analyse der DNA-Methylierung, das Einblick in die Auflösung auf Einzelbasenebene bietet.
DNA 6mA Sequenzierung
Die DNA 6mA-Sequenzierung konzentriert sich auf die Erkennung und Quantifizierung von N6-Methyladenin (6mA) in DNA, einem aufkommenden epigenetischen Marker.
DAP-Seq-Dienst
DNA-Affinitätsreinigung-Sequenzierung (DAP-Seq) ist ein Verfahren, das eingesetzt wird, um Wechselwirkungen zwischen Proteinen und DNA aufzudecken, und somit unser Verständnis von DNA-bindenden Proteinen und deren Funktionen in der Genregulation zu verbessern.
oxBS-seq
Oxidative Bisulfite-Sequenzierung (oxBS-seq) ist eine spezialisierte Methode, die verwendet wird, um zwischen 5-Methylcytosin (5mC) und 5-Hydroxymethylcytosin (5hmC) in DNA zu unterscheiden und deren unterschiedliche Rollen in der epigenetischen Regulation zu beleuchten.
5mC/5hmC-Sequenzierung
Unser Hochdurchsatz-5mC/5hmC-Sequenzierungsdienst nutzt mehrere ausgereifte und stabile Plattformen mit hoher Effizienz, Einfachheit und Genauigkeit, um Ihre epigenetische Forschung zu unterstützen.
Unsere RNA-Methylierungslösungen einschließen:
MeRIP-Sequenzierung (m6A-Analyse)
MeRIP-Sequenzierung konzentriert sich auf RNA-Modifikationen, insbesondere N6-Methyladenosin (m6A), und hilft, das posttranskriptionale Regulation und die Dynamik der RNA-Modifikationen zu verstehen.
RIP-Seq
RNA-Immunopräzipitation-Sequenzierung (RIP-Seq) wird eingesetzt, um Interaktionen zwischen RNA und Proteinen zu untersuchen, und bietet wertvolle Einblicke in die Funktionen von RNA-bindenden Proteinen und deren Einfluss auf die RNA-Funktionalität.
2'-O-RNA-Methylierungs-Sequenzierung
Der 2'-O-RNA-Methylierungs-Sequenzierungsdienst (RiboMeth-seq) bietet eine umfassende Erkennung von 2'-O-RNA-Methylierungsmodifikationen über eine Vielzahl von RNA-Molekülen, einschließlich snoRNA, tRNA und rRNA. RiboMeth-seq nutzt hauptsächlich entweder alkalische oder Metallionenbehandlungen, um zufällige RNA-Spaltungen zu induzieren. Positionen der 2'-O-Methylierung zeigen eine Resistenz gegen Spaltung und bleiben somit intakt; im Gegensatz dazu sind nicht-methylierte Stellen anfällig für Spaltung oder Abbau. Durch die Identifizierung der Stellen, die der Spaltung widerstehen, kartiert RiboMeth-seq effektiv die 2'-O-Methylierungsstellen.
Nanopore-RNA-Methylierungs-Sequenzierung
Nanopore-RNA-Methylierungssequenzierung ist ein Verfahren, das Nanopore-Technologie verwendet, um RNA-Moleküle direkt zu sequenzieren und ihre Methylierungsmodifikationen zu erkennen. Diese Technik ermöglicht eine präzise Lokalisierung und Identifizierung von Methylierungsstellen durch die Echtzeitanalyse von Änderungen elektrischer Signale, während RNA-Moleküle durch das Nanopore passieren. Mit den Vorteilen der direkten Sequenzierung und langen Lesegrößen dient diese Technologie als leistungsstarkes Werkzeug zur Untersuchung der Rolle von RNA-Methylierung in der Genexpression und Zellfunktion.
Vorteile der Epigenom-Sequenzierung
- Umfassende EinblickeDie Epigenomforschung bietet eine umfassende Übersicht über epigenetische Modifikationen im gesamten Genom, einschließlich DNA-Methylierung, Histonmodifikationen und der regulatorischen Rollen von nicht-kodierenden RNAs. Dieser ganzheitliche Ansatz ist entscheidend für das Verständnis komplexer genetischer Regulationsnetzwerke und ihrer dynamischen Reaktionen.
- Sensitivität und SpezifitätModerne epigenomische Techniken, wie ChIP-seq, Bisulfid-Sequenzierung, und ATAC-seqbieten außergewöhnliche Sensitivität und Spezifität. Diese Methoden ermöglichen die präzise Erkennung und Quantifizierung subtiler Veränderungen in epigenetischen Markierungen, die entscheidend sind für das Verständnis zellulärer Prozesse und Krankheitsmechanismen auf molekularer Ebene.
- HochdurchsatzanalyseEpigenomische Methoden ermöglichen die Hochdurchsatzanalyse von Tausenden von genomischen Regionen und epigenetischen Loci gleichzeitig. Diese Fähigkeit verbessert die Forschungseffizienz erheblich und unterstützt großangelegte Untersuchungen biologischer Systeme und komplexer genetischer Interaktionen.
- Zeitliche und räumliche AuflösungFortgeschrittene epigenomische Werkzeuge bieten zeitliche und räumliche Auflösung epigenetischer Modifikationen und erfassen dynamische Veränderungen über verschiedene Entwicklungsstadien und Zelltypen hinweg. Diese detaillierte Einsicht in die epigenetische Dynamik trägt entscheidend zum Verständnis der Entwicklungsbiologie und der Pathogenese von Krankheiten bei.
Anwendung der Epigenom-Sequenzierung
- KrebsforschungDie Epigenomik findet umfangreiche Anwendung in der Krebsforschung, wo die Analyse epigenetischer Veränderungen in Tumorzellen potenzielle Biomarker und therapeutische Ziele identifiziert. Dies trägt erheblich zur Weiterentwicklung personalisierter Behandlungsstrategien bei.
- EntwicklungsbiologieDas Studium epigenetischer Regulationsmechanismen während der embryonalen Entwicklung verbessert unser Verständnis der zellulären Differenzierung und Organogenese auf molekularer Ebene.
- NeurowissenschaftenDie Erforschung der Rolle epigenetischer Modifikationen in der Neuroentwicklung, synaptischen Plastizität und neurodegenerativen Erkrankungen offenbart Mechanismen, die neurologische Störungen zugrunde liegen.
- UmweltwissenschaftenDie Epigenomik wird verwendet, um Umwelteinflüsse wie Ernährung, Schadstoffe und Lebensstil auf die Genexpression und epigenetische Modifikationen zu untersuchen. Dies hilft, die Beziehung zwischen Umwelt und Gesundheit zu bewerten.
- PflanzenwissenschaftenIn der epigenetischen Forschung an Pflanzen deckt die Epigenomik molekulare Mechanismen auf, die das Wachstum, die Entwicklung und die Reaktionen von Pflanzen auf Umweltstressoren steuern, was die Verbesserung von Nutzpflanzen und die landwirtschaftliche Produktivität erleichtert.
Musteranforderungen
Unten finden Sie die Beispielanforderungen für einige unserer epigenomischen Sequenzierungsdienste. Für detailliertere Informationen besuchen Sie bitte die spezifischen Dienstleistungsseiten. Wenn Sie an unseren Dienstleistungen interessiert sind, bitte Kontaktieren Sie uns um die Anforderungen an die Sequenzierungsproben zu bestätigen.
Tabelle 1. Epigenomik-Sequenzierung
| Dienstleistung | Probenart | Empfohlene Menge | Mindestmenge | Mindestkonzentration |
|---|---|---|---|---|
| MeDIP-Seq/hMeDIP-seq | Genomische DNA | ≥ 2 µg | 1 µg | 20 ng/μL |
| WGBS (Whole Genome Bisulfit-Sequenzierung) Sequenzierung |
Genomische DNA Zelle Gewebe |
≥ 1 μg ≥ 1 x106 ≥ 50 mg |
200 ng |
10 ng/µL |
| RRBS (Reduzierte Repräsentations-Bisulfid) Sequenzierung) |
Genomische DNA Zelle Gewebe |
≥ 1 μg ≥ 5 x106 ≥ 30 mg |
20 ng 3×103 |
20 ng/µL |
| Gezielte Bisulfid-Sequenzierung | Genomische DNA Zelle Gewebe |
≥ 500 ng ≥ 1 x106 ≥ 20 mg |
50 ng | 10 ng/µL |
| oxWGBS-Seq | Genomisches DNA | ≥ 3 µg | 1 µg | 30 ng/µL |
| oxRRBS-Seq | Genomische DNA | ≥ 2 µg | 50 ng/μL | |
| oxTBS-Seq | Genomisches DNA | ≥ 1 µg | 20 ng/μL | |
| Epityper | Genomisches DN | ≥ 1 µg | ||
| DNA 6mA-IP-Seq | Genomische DNA | ≥ 5 µg | 20 ng/μL | |
| ChIP-seq | ChIPed DNA Zelle Gewebe |
≥ 10 ng ≥ 2 x 107 ≥ 500 mg |
5 ng 1×105 |
1 ng/µL |
| DAP-seq | TF Zelle Gewebe |
≥ 5 µg ≥ 5 x106 ≥ 500 mg |
1 µg 200 mg |
20 ng/µL |
| ATAC-seq | Zelle Gewebe |
≥ 1 x106 ≥ 500 mg |
5×104 200 mg |
1 ng/µL |
Tabelle 2. RNA-Epigenomik-Sequenzierung
| Dienstleistung | Probenart | Empfohlene Menge | Mindestmenge | Mindestkonzentration |
|---|---|---|---|---|
| RIP-seq | IPed RNA Zelle Gewebe |
≥ 100 ng ≥ 5×107 ≥ 500 mg |
40 ng 200 mg |
5 ng/μL |
| eCLIP-Seq | IPed RNA Zelle Gewebe |
≥ 100 ng ≥ 3×107 ≥ 500 mg |
40 ng 200 mg |
5 ng/μL |
| MeRIP (Methyliertes RNA) Immunopräzipitation |
Gesamt-RNA Zelle Gewebe |
≥ 10 μg ≥ 1×107 ≥ 500 mg |
2 μg 200 ng |
1 ng/µL |
| RNA-BS-Seq (RNA-Bisulfid-Sequenzierung) | Gesamt-RNA Zelle Gewebe |
≥ 10 μg ≥ 1×107 ≥ 500 mg |
100 mg |
Analyse-Pipeline
Der Analyse-Workflow für Sequenzierungsdaten in der Epigenomik folgt im Allgemeinen dem unten dargestellten Schema. Da jede Methode spezifische Schritte umfasst, können Sie auf die jeweiligen Abschnitte für detaillierte Verfahren zur Datenanalyse verweisen.
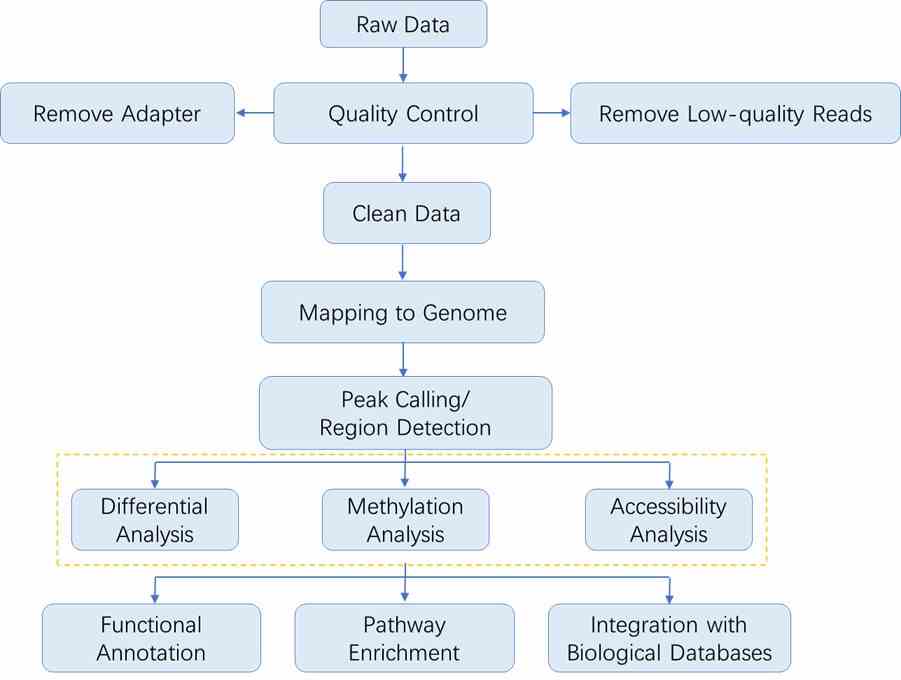
Ergebnisse anzeigen
Im Folgenden finden Sie eine teilweise Präsentation der Ergebnisse unserer epigenomischen Sequierungsanalysen. Für detailliertere Informationen besuchen Sie bitte die entsprechenden Serviceseiten.
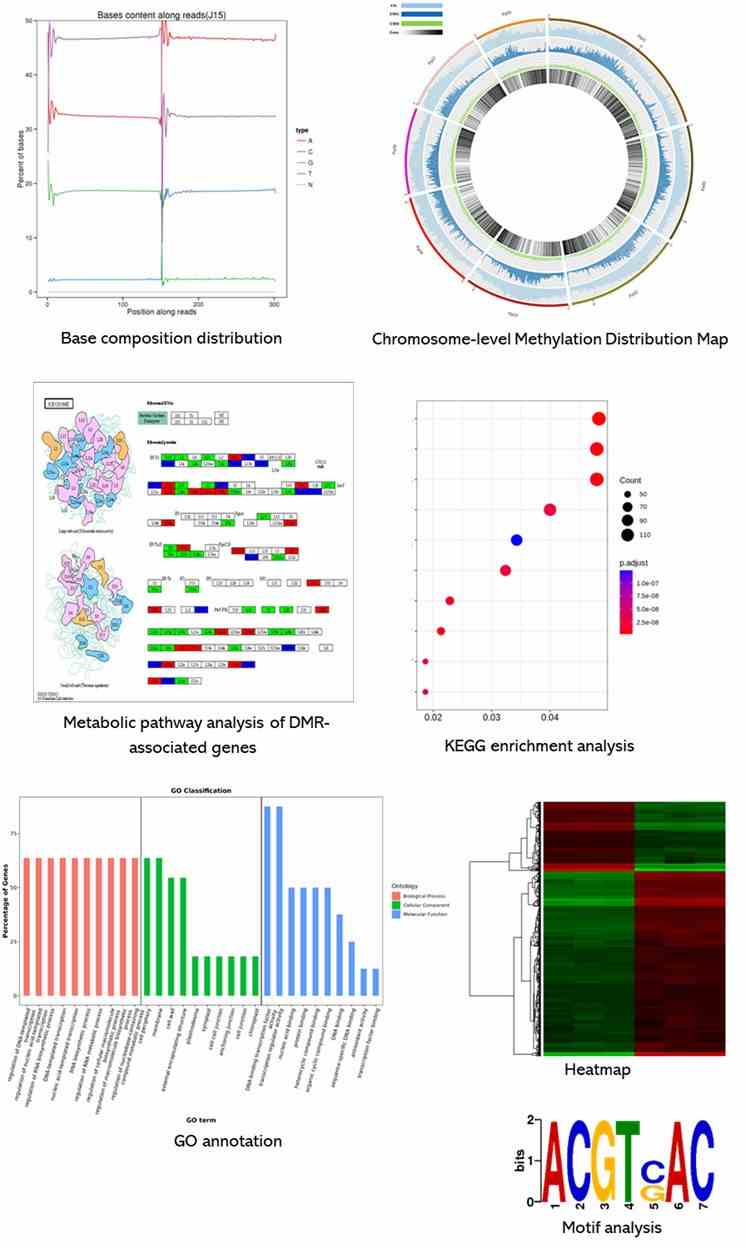
Referenzen
- Jeong H.M., u. a.Effizienz der immunpräzipitierten Bisulfit-Sequenzierung von methyliertem DNA für die Analyse der DNA-Methylierung im gesamten Genom. Epigenomik. 2016, 8(8):1061-77.
- Roundtree I A, Evans M E, Pan T, et al. Dynamische RNA-Modifikationen in der Regulierung der Genexpression. Zelle, 2017, 169(7):1187-1200.
Disruption of tRNA biogenesis enhances proteostatic resilience, improves later-life health, and promotes longevity
Plos Biology | 2024Methylation in the CHH Context Allows to Predict Recombination in Rice
International Journal of Molecular Sciences | 2022High-Fat Diets Fed during Pregnancy Cause Changes to Pancreatic Tissue DNA Methylation and Protein Expression in the Offspring: A Multi-Omics Approach
International Journal of Molecular Sciences | 2024Folate Carrier Deficiency Drives Differential Methylation and Enhanced Cellular Potency in the Neural Plate Border
Frontiers in Cell and Developmental Biology | 2022

