
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
EM-seq Dienst
DNA-Methylierung ist entscheidend für die Regulierung der Genexpression und beeinflusst eine Vielzahl biologischer Prozesse, einschließlich Entwicklung, Alterung und verschiedener Krankheiten wie Krebs. Mit dem Fortschritt der Genomforschung wächst die Nachfrage nach effizienteren und genaueren Methoden zur DNA-Methylierungserkennung. EM-seq (enzymatische Methylierungssequenzierung) tritt als neuartige Technologie zur Analyse der DNA-Methylierung hervor und führt die präzise Methylierungsanalyse mit modernsten Vorteilen an. Es bietet beispiellose Lösungen mit erhöhter Sensitivität, Genauigkeit und minimalen Anforderungen an die DNA-Menge.
Bei CD Genomics nutzen wir die Leistungsfähigkeit der EM-seq-Technologie, um außergewöhnliche Methylierungsanalysedienste anzubieten, die es Ihnen ermöglichen, die Feinheiten der Genregulation und die Mechanismen, die verschiedenen Krankheiten zugrunde liegen, zu erforschen. Auf dieser umfassenden Serviceseite werden wir die wichtigsten Aspekte von EM-seq, seine technologischen Vorteile, Anwendungsgebiete und die Art und Weise, wie wir fortschrittliche Unterstützung bieten, die auf Ihre Forschungsbedürfnisse zugeschnitten ist, näher beleuchten.
Einführung in die EM-seq-Technologie
EM-seq ist eine enzymbasierte DNA-Methylierungssequenzierungstechnologie, die zwischen unmethylierter Cytosin (C) und methylierter 5-Methylcytosin (5mC) mithilfe enzymatischer Methoden unterscheidet. Im Gegensatz zur traditionellen Bisulfid-Konversionsmethode (WGBS) verwendet EM-seq enzymatische Katalyse, um unmethylierte Cytosine in Uracil (U) umzuwandeln, während methylierte Cytosine unverändert bleiben. Während der anschließenden Amplifikation und Sequenzierung wird Uracil als Thymin (T) erkannt. Dieser Ansatz ermöglicht eine präzise Erkennung von Methylierungsstellen.
Vergleich der enzymatischen Umwandlung und der Bisulfitumwandlung
Die konventionelle Bisulfit-Konversionsmethode ist effektiv zur Erkennung von Methylierung. Sie erfordert jedoch strenge Reaktionsbedingungen, die oft DNA schädigen, was ihre Anwendung bei DNA-Proben mit niedrigem Input einschränkt. EM-seq überwindet diese Probleme, indem es die DNA-Integrität bewahrt und es ermöglicht, eine breitere Palette von Proben zu verarbeiten, einschließlich spuren- und fragiler DNA-Proben wie zirkulierendem Tumor-DNA (ctDNA).
Experimentelles Prinzip
EM-seq bietet eine umfassende und robuste Lösung zur Identifizierung von Methylierungsregionen im menschlichen Genom. Während der Bibliotheksvorbereitung wird eine einzigartige enzymatische Umwandlung eingesetzt, die viel weniger Schäden an der DNA verursacht und kleinere Probenmengen erfordert, was zu hochwertigeren und leistungsfähigeren Bibliotheken führt. Das maßgeschneiderte Methylierungsproben-Design von Twist bietet effektive, spezialisierte Sonden für die gezielte CpG-Anreicherung. Optimierte Hybridisierungsreagenzien erhöhen die Flexibilität der Arbeitsabläufe und verbessern die On-Target-Raten.
Die Methylierungssequenzierung umfasst enzymatische oder chemische Methoden, die nicht-methylierte Cytosine durch Deaminierung in Uracil umwandeln, während methylierte Cytosine intakt bleiben. Während der Amplifikation paart sich komplementäres Adenin mit Uracil auf dem komplementären Strang, wodurch Thymidin an der ursprünglichen nicht-methylierten Cytosin-Position eingeführt wird. Das Endprodukt der Sequenz ist asymmetrisch und erzeugt zwei unterschiedliche doppelsträngige DNA-Moleküle nach der Umwandlung. Bei methylierter DNA führt der Prozess zu einem alternativen Sequenzmuster, das den Unterschied zwischen methylierten und nicht-methylierten Regionen in DNA-Sequenzen veranschaulicht.
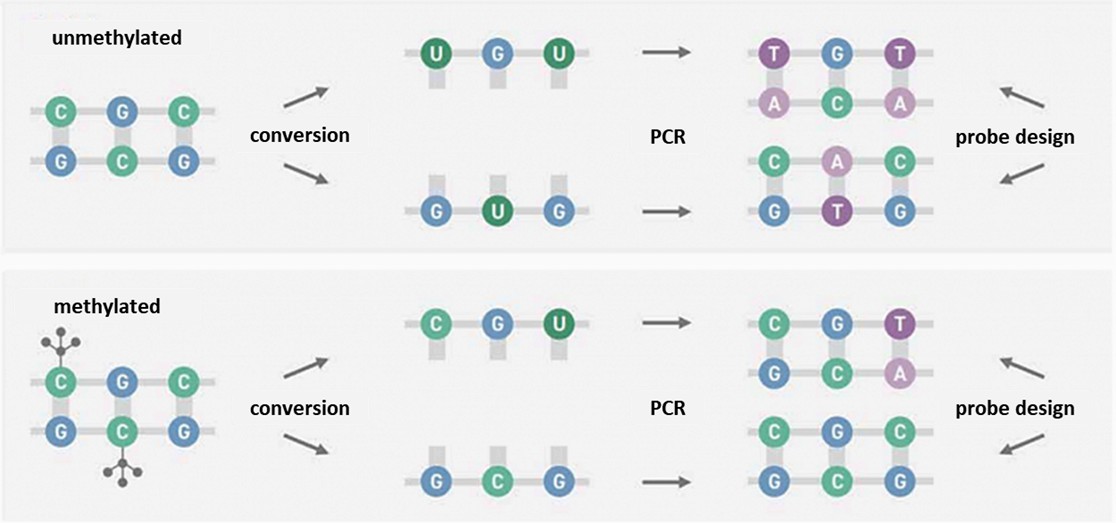 Abbildung 1. Die Methylierungssequenzierung umfasst enzymatische oder chemische Methoden.
Abbildung 1. Die Methylierungssequenzierung umfasst enzymatische oder chemische Methoden.
In der Anfangsphase der Reaktion spielt die Ten-Eleven Translocation Dioxygenase 2 (TET2) eine entscheidende Rolle bei der Umwandlung von methylierte Cytosinen wie 5-Methylcytosin (5mC) und 5-Hydroxymethylcytosin (5hmC) in 5-Carboxycytosin (5caC). Dies wird durch einen Oxidationsbooster, 5-Glucosylhydroxymethylcytosin (5ghmC), weiter verstärkt. Diese Reaktionen dienen als Schutzmechanismen, die 5mC und 5hmC vor nachfolgenden Deaminierungsaktivitäten abschirmen.
Vor der Deaminierung von Cytosinen zu Uracil durch das Enzym APOBEC wird die DNA denaturiert, um sie auf weitere Reaktionen vorzubereiten. Die anschließende Polymerase-Kettenreaktion (PCR) wandelt modifiziertes 5mC oder 5hmC in Cytosin um und konvertiert Uracil in Thymin. Nach der PCR spiegelt die Nukleinsäuresequenz die der bisulfidkonvertierten Sequenzen wider, was die Kompatibilität von EM-seq mit bestehenden Analyse-Workflows, einschließlich Tools wie Bismark und bwa-meth, gewährleistet.
Effizienz der enzymatischen Umwandlung
| Metrisch | Unmethyliertes Lambda-DNA | CpG-methylierte pUC19-DNA |
|---|---|---|
| Erwartete Umwandlungseffizienz | ≥99,5% | ≥99,5% |
| Gemessene Umwandlungseffizienz | 99,77 % | 99,57 % |
| Erwartetes CpG-Methylierungsniveau | ~0,5% | 95-98% |
| Gemessener CpG-Methylierungsgrad | 0,22228% | 95,7572 % |
Vorteile von EM-seq
EM-seq übertrifft die traditionelle Bisulfit-Konversionsmethode in mehreren wichtigen Aspekten, insbesondere bei der Arbeit mit geringen Mengen DNA. Hier sind einige Hauptvorteile der EM-seq-Technologie:
1. Niedriger DNA-Eingang und hohe EmpfindlichkeitEiner der Hauptvorteile von EM-seq ist die Fähigkeit, hochwertige Sequenzierungsdaten aus minimalen DNA-Mengen zu erhalten. Im Gegensatz zu WGBS verursacht EM-seq weniger Schäden an der DNA, wodurch eine Sequenzierung mit nur 10 ng DNA möglich ist. Dieses Merkmal ist von unschätzbarem Wert für die Analyse seltener oder kostbarer Proben, wie z.B. zirkulierendes Tumor-DNA (ctDNA) im Plasma, die typischerweise niedrig in Konzentration und Molekulargewicht sind.
2. Längere, vollständigere BibliotheksfragmenteIm Vergleich zu WGBS erzeugt EM-seq längere und intaktere DNA-Fragmente, wodurch Sequenzierungslücken minimiert werden. Dies erhöht die Vollständigkeit der genomweiten Methylierungsinformationen, insbesondere in schwer zu sequenzierenden Regionen, indem es effektiv die durch kurze DNA-Fragmente verursachten Sequenzierungsblindstellen reduziert.
3. Überlegene GC-AbdeckungsuniformitätDNA-Regionen mit hohem GC-Gehalt stellen oft Herausforderungen bei der Sequenzierung dar, da potenzielle Verzerrungen die Genauigkeit beeinträchtigen können. EM-seq zeichnet sich in diesem Bereich aus und gewährleistet eine gleichmäßige Abdeckung sowohl in GC-reichen als auch in AT-reichen Bereichen, wodurch die häufige GC-Abdeckungsverzerrung in WGBS vermieden wird. Diese Eigenschaft ist entscheidend für die genaue Analyse von Methylierungszuständen im gesamten Genom.
4. Genauere Erkennung von CpG-InselnEM-seq ist besonders effektiv bei der Erkennung von CpG-Inseln. Bei vergleichbaren Sequenzierungstiefen kann EM-seq mehr CpG-Stellen aufdecken, von denen viele durch traditionelle WGBS-Methoden übersehen werden könnten. Diese Fähigkeit ist besonders bedeutend für das frühe Screening von Krankheiten wie Krebs, bei dem Methylierungsänderungen in CpG-Inseln als frühe Krankheitsmarker dienen können.
Anwendungen von EM-seq
In der heutigen schnell fortschreitenden wissenschaftlichen Landschaft hat sich die EM-seq-Technologie als leistungsstarkes Werkzeug mit umfangreichen Anwendungen in verschiedenen Bereichen etabliert. Ihre Fähigkeit, DNA-Proben in niedrigen Konzentrationen zu detektieren und Veränderungen in der DNA-Methylierung zu untersuchen, macht sie besonders wertvoll. So transformiert EM-seq wichtige Forschungs- und Entwicklungsbereiche:
- Früherkennung von Krebs: Screening und FlüssigbiopsienEM-seq spielt eine entscheidende Rolle bei der frühen Erkennung und fortlaufenden Überwachung von Krankheiten, indem es cfDNA-Methylierungsmarker in Körperflüssigkeiten wie Plasma und Urin identifiziert.
- Eintauchen in die EpigenetikDiese Technik hilft dabei, zu entdecken, wie die DNA-Methylierung die Genexpression und biologische Prozesse beeinflusst und beleuchtet die epigenetischen Variationen in verschiedenen biologischen Zuständen.
- Präzision in der Analyse von SpurenprobenOb es sich um wertvolle Proben wie Eizellen, Spermatozyten oder Embryonen handelt, EM-seq liefert hochpräzise Methylierungsinformationen, selbst aus winzigen Proben.
- Pionierhafte Entdeckung von BiomarkernEM-seq ist entscheidend für die Auswahl und Validierung von Methylierungsmarkern, die mit spezifischen biologischen Prozessen in Verbindung stehen, und unterstützt zukünftige Forschungs- und Entwicklungsbemühungen.
- Genomische und epigenetische Einblicke in KrankheitenDurch die Entschlüsselung der Rolle der DNA-Methylierung trägt EM-seq erheblich zu unserem Verständnis biologischer Mechanismen bei und liefert Einblicke in komplexe Krankheitsprozesse.
- Verbesserung der ArzneimittelentwicklungDie Technologie wird genutzt, um zu untersuchen, wie Medikamente die DNA-Methylierungsmuster beeinflussen, und bietet wichtige molekulare Daten, die das Screening und die Optimierung von Arzneimitteln erleichtern.
- Erforschung der Biodiversität und EvolutionEM-seq analysiert Methylierungsmuster über verschiedene Arten oder Populationen hinweg und hilft dabei, adaptive Veränderungen und die genetischen Mechanismen, die sie steuern, aufzudecken.
- Analyse der Methylierung des TranskriptionsstartortsEs bewertet genau den Methylierungsstatus an Transkriptionsstartstellen (TSS), insbesondere in GC-reichen Regionen, um deren regulatorische Rollen in der Genexpression offenzulegen.
Plattformvergleich für die DNA-Methylierungsanalyse
| Projektkategorie | Plattform | Funktionen | Datenvolumen | Loci (10X) | Anfangsbetrag (μg) | Technisches Prinzip | Musteranforderung (Empfohlen) |
|---|---|---|---|---|---|---|---|
| 850.000 | Chip | Einzelbasisauflösung | / | 860.000 | 0,25 | Bisulfit-Konversion | Vielfache von 8 |
| WGBS | Hochdurchsatz | Einzelbasenauflösung | 90G | 5 Millionen | 1 | Bisulfit-Konversion | / |
| MC-seq | Hochdurchsatz | Einzelbasenauflösung | 20G | 2,7 Millionen (84M) | 1 | Bisulfit-Konversion | / |
| EM-seq | Hochdurchsatz | Einzelbasenauflösung | 25G | 4 Millionen (134M) | 0,01 | Enzymatisch | Vielfache von 8 |
| scWGBS | Hochdurchsatz | Einzelbasenauflösung | 15G | 5 Millionen | 0,01 | Bisulfit-Konversion | / |
| Pyrosequenzierung | Erstgen. | Einzelbasisauflösung | 50 - 90 bp | / | 0,5 | Bisulfit-Konversion | / |
EM-seq Arbeitsablauf
Die Inanspruchnahme unserer Dienstleistungen bietet ein nahtloses Erlebnis, das darauf ausgelegt ist, hochwertige Ergebnisse für Ihre EM-seq-Projekte zu liefern. So stellen wir an jedem Schritt Exzellenz sicher:
Schritt 1: DNA-ExtraktionWir beginnen mit der sorgfältigen Extraktion von hochwertiger genomischer DNA aus Ihren Proben. Dieser entscheidende erste Schritt stellt sicher, dass die DNA intakt und frei von Verunreinigungen ist, und bildet eine solide Grundlage für weitere Analysen.
Schritt 2: BibliotheksvorbereitungMit modernsten Technologien zur Bibliotheksvorbereitung bereiten wir Ihre DNA-Proben geschickt für die EM-seq-Sequenzierung vor. Dieser Prozess ist entscheidend, um sicherzustellen, dass Ihre Proben optimal für genaue Ergebnisse vorbereitet sind.
Schritt 3: SequenzierungIhre DNA-Bibliothek wird dann mit den neuesten Technologien der nächsten Generation für die Sequenzierung (NGS) sequenziert. Diese Phase ermöglicht es uns, umfassende genomweite Methylierungsdaten zu erfassen und bietet einen umfassenden Überblick über die epigenetische Landschaft.
Schritt 4: DatenanalyseUnser Expertenteam für Bioinformatik erstellt einen detaillierten Datenanalysebericht für Sie. Dieser Bericht umfasst Methylierungslevels, die Identifizierung von unterschiedlich methylierten Regionen und die Erkennung von CpG-Inseln, sodass Sie umsetzbare Erkenntnisse aus Ihren Daten gewinnen können.
 Abbildung 1. Methylierungssequenzierung umfasst enzymatische oder chemische Methoden.
Abbildung 1. Methylierungssequenzierung umfasst enzymatische oder chemische Methoden.
Dienstspezifikation
Beispielanforderungen
|
|
 Klicken |
Sequenzierungsstrategien
|
 |
Bioinformatikanalyse Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an: 1. Genom-Ausrichtungsstatistiken 2. Gesamte Bewertung des Methylierungsniveaus
|
Analyse-Pipeline

Liefergegenstände
- Originale Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Detaillierte Methylierungsprofilierung
EM-seq, als revolutionäre DNA-Methylierungssequenzierungstechnologie, übertrifft die traditionelle Bisulfit-Konversionsmethode mit ihren Vorteilen in Bezug auf niedrigen DNA-Eingang, hohe Sensitivität und gleichmäßige GC-Abdeckung und bietet hochwertigere und genauere Methylierungsdaten für verschiedene Forschungsbereiche. Bei CD Genomics setzen wir uns dafür ein, unseren Kunden die fortschrittlichsten EM-seq-Dienste anzubieten, um Ihnen bei der eingehenden Erforschung der Genregulation, der Krankheitsmechanismen, der Krebsforschung und anderer Bereiche zu helfen.
Egal, ob Sie Grundlagenforschung betreiben oder fortschrittliche Diagnosetools entwickeln, EM-seq-Dienste können hervorragende Unterstützung für Ihre Forschung bieten. Zögern Sie nicht, uns zu kontaktieren, um mehr über EM-seq zu erfahren oder ein Angebot anzufordern, und lassen Sie diese fortschrittliche Technologie Ihnen helfen, neue wissenschaftliche Durchbrüche zu erzielen.
Demo-Ergebnisse
Teilweise Ergebnisse sind unten aufgeführt:
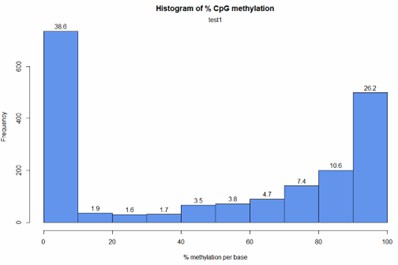
CpG-Methylierungsverteilungshistogramm
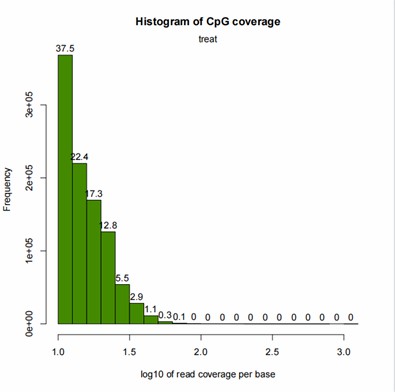
CpG-Abdeckungs-Histogramm
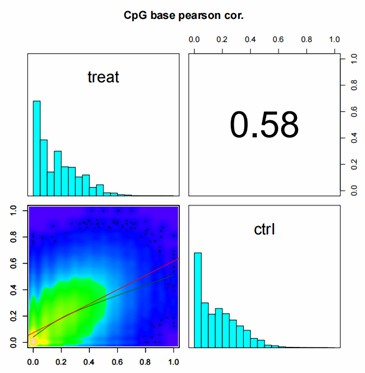
Korrelationsanalyse zwischen Proben
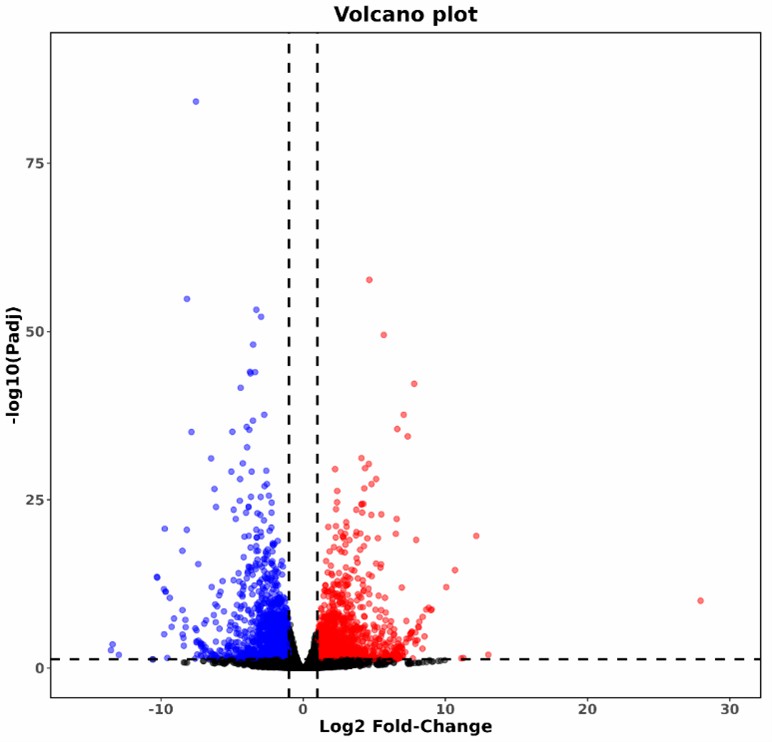
Vulkan-Plot von differentiell methylierten Stellen
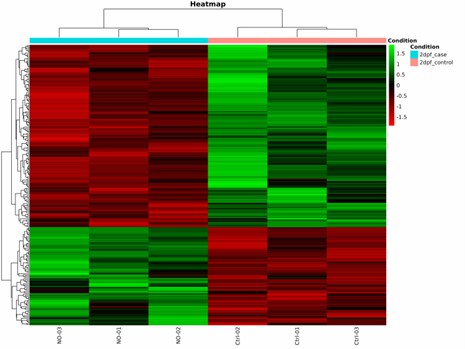
Heatmap der unterschiedlich methylierten Stellen

KEGG-Anreicherungsanalyse von unterschiedlich methylierten Regionen
EM-seq häufig gestellte Fragen (FAQs)
Was sollte beim Extrahieren von gDNA oder cfDNA beachtet werden?
Beim Extrahieren von genomischer DNA (gDNA) oder zellfreier DNA (cfDNA) wird empfohlen, Folgendes zu erreichen:
- gDNA-AnforderungGesamt ≥20 ng, Konzentration ≥2 ng/μl
- cfDNA-AnforderungGesamt ≥20 ng, Konzentration ≥2 ng/μl
Zusätzlich ist es entscheidend, EDTA oder EB nicht als Lösungsmittel für Ihr extrahiertes gDNA/cfDNA zu verwenden, da diese Komponenten die enzymatische Effizienz negativ beeinflussen können.
Welche Vorsichtsmaßnahmen sollten beim Trennen von Serum oder Plasma getroffen werden?
Beim Trennen von Serum oder Plasma:
- Vermeiden Sie das Einfrieren und AuftauenVollblutproben sollten keinen Frost-Tau-Zyklen unterzogen werden.
- Rechtzeitige BearbeitungBereiten Sie Plasma oder Serum so schnell wie möglich nach der Blutentnahme vor.
- LagerbedingungenSerum oder Plasma können bei -80°C gelagert werden, um die Integrität zu erhalten. Wiederholte Gefrier-Auftau-Zyklen sollten vermieden werden, um die Probenqualität zu bewahren.
Wie wird die Konversionsrate berechnet?
Während der Vorbereitung der Methylierungsbibliothek geht die theoretische Umwandlung davon aus, dass alle unmethylierte Cytosine (C) in Thymin (T) umgewandelt werden (zunächst in Uracil (U) umgewandelt, dann während der PCR in T). Allerdings können nicht alle Stellen umgewandelt werden, und der Methylierungsstatus der DNA ist anfänglich unbekannt. Daher können die tatsächlichen Umwandlungsraten nicht direkt aus der Proben-DNA bestimmt werden.
Um dies zu adressieren, wird lambda-DNA (Bakteriophagen-DNA), die nur unmethylierte Cytosine enthält, als negativer Kontrollstandard mit bekanntem Methylierungsstatus verwendet. Durch die Einbeziehung von lambda-DNA in die Bibliotheksvorbereitung, Verarbeitung und Sequenzierung zusammen mit der Proben-DNA wird die Umwandlungsrate von lambda-DNA berechnet, um als Indikator für die Gesamtumwandlungsrate der Probe zu dienen.
EM-seq Fallstudien
Multimodale epigenetische Sequenzierungsanalyse (MESA) von zellfreier DNA zur nicht-invasiven Krebsdetektion
Journal: Genommedizin
Impact-Faktor: 7,324
Veröffentlicht: 16. Januar 2024
Hintergrund
Die Methylierung von zellfreier DNA (cfDNA) hat sich als vielversprechender Biomarker für die frühe Krebsdiagnose etabliert und überwindet die Einschränkungen genetischer Veränderungsansätze. Die enzymatische Methyl-sequenzierung (EM-seq) verbessert die traditionelle Bisulfit-Sequenzierung, indem sie die DNA-Integrität bewahrt und multimodale epigenetische Analysen ermöglicht, die die Genauigkeit der Krebsdiagnose erhöhen.
Materialien & Methoden
- Vollblutprobe
- Klärendes Plasma
- Gezielte Sequenzierung
- Gezielte EM-seq
- Datenverarbeitung und Qualitätskontrolle
- Multimodale Merkmalsextraktion
- Merkmalsauswahl
- Kreuzkohortenvalidierungsanalyse
Ergebnisse
MESA (Multimodale Epigenetische Sequenzierungsanalyse) zeigte eine starke Leistung bei der Erkennung von kolorektalem Krebs durch die Integration mehrerer epigenetischer Merkmale aus cfDNA. Zu den wichtigsten Ergebnissen gehören:
1. Methylierungsanalyse:
- cfDNA-Methylierung allein unterschied Krebs- von Nicht-Krebs-Proben mit hoher Genauigkeit (AUC = 0,8663 für Kohorte 1 und AUC = 0,8293 für Kohorte 2).
- Krebsmuster zeigten höhere Methylierungsniveaus an den Ziel-CpG-Stellen, was mit einer Promotorhypermethylierung in Tumoren übereinstimmt.
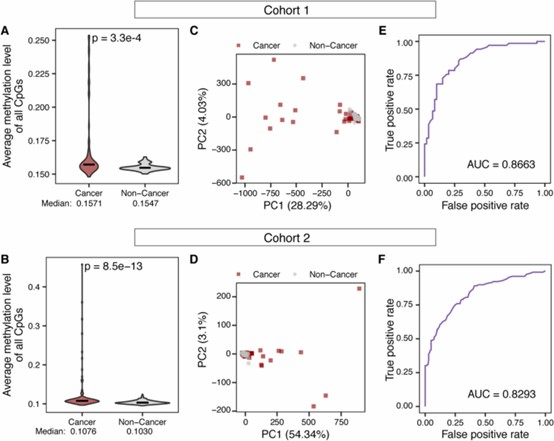 Differenzielle cfDNA-Methylierung zwischen Krebs- und Nicht-Krebs-Proben ermöglicht eine genaue Krebsdiagnose.
Differenzielle cfDNA-Methylierung zwischen Krebs- und Nicht-Krebs-Proben ermöglicht eine genaue Krebsdiagnose.
2. Einblicke in die Nucleosomenorganisation:
- MESA erfasste effektiv die Nukleosomenbesetzung und -unschärfe und offenbarte wichtige strukturelle Unterschiede zwischen Krebs- und Normalproben.
- Polyadenylierungsstellen wurden erstmals in die Analyse einbezogen, was neuartige Einblicke in die krebsbezogene Chromatinorganisation bietet.
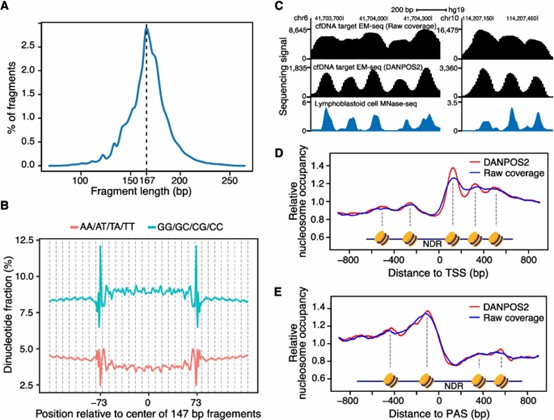 Nukleosomenorganisationsinformationen aus gezieltem EM-seq von cfDNA.
Nukleosomenorganisationsinformationen aus gezieltem EM-seq von cfDNA.
3. Krebsdiagnose anhand von Nucleosomenmerkmalen:
- Die Nucleosombelegung allein erreichte AUCs von 0,8494 (Kohorte 1) und 0,9213 (Kohorte 2).
- Die Einbeziehung von Daten zu Polyadenylierungsstellen verbesserte die Genauigkeit der Krebsdiagnose weiter.
- Die Nucleosomen-Unschärfe, eine neuartige Kennzahl zur Erfassung der Chromatin-Heterogenität, bot zusätzliche Vorhersagekraft.
4. Multimodale Integration für verbesserte Genauigkeit:
- Die Kombination von Methylierung, Nukleosomenbesetzung, Unschärfe und WPS verbesserte die Klassifikationsleistung.
- Das multimodale Modell übertraf konsequent Modelle mit einzelnen Merkmalen in verschiedenen Krebsstadien und Kohorten.
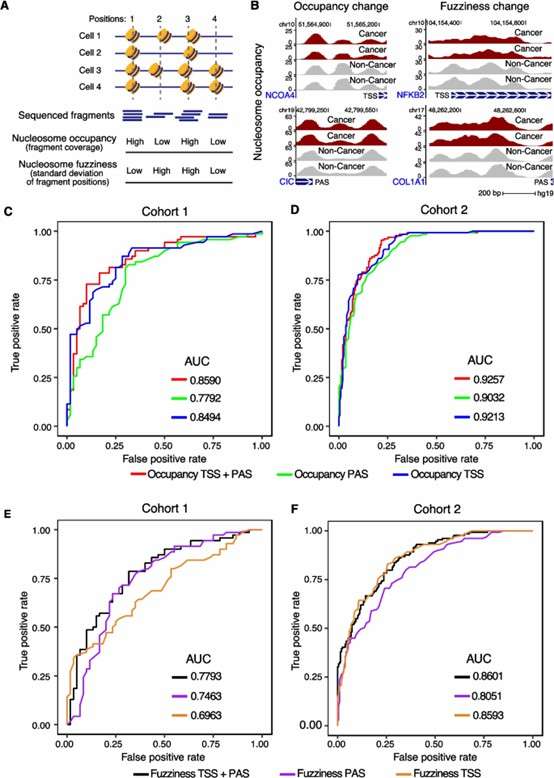 Genauigkeit der Krebsdetektion basierend auf Nukleosomenbelegung und Unschärfe.
Genauigkeit der Krebsdetektion basierend auf Nukleosomenbelegung und Unschärfe.
5. Kreuzvalidierung zwischen Kohorten:
- MESA war in verschiedenen Kohorten robust und hielt eine hohe Vorhersagegenauigkeit aufrecht.
- Der Ansatz war anpassungsfähig an andere bisulfidfreie Sequenzierungsmethoden (z. B. cfDNA TAPS) und war effektiv bei der Erkennung von hepatozellulären und pankreatischen Krebsarten.
Fazit
MESA integriert multiple epigenetische Modalitäten und verbessert die Erkennungsgenauigkeit für kolorektale, Leber- und Bauchspeicheldrüsenkrebs, validiert in vier Kohorten. Es stellt einen bedeutenden Durchbruch in der nicht-invasiven Krebsdiagnose dar, indem es umfassende epigenetische Profile von cfDNA nutzt.
Referenz:
- Li, Y., Xu, J., Chen, C. u. a.Multimodale epigenetische Sequenzierungsanalyse (MESA) von zellfreier DNA zur nicht-invasiven Erkennung von kolorektalem Krebs. Genommedizin 16, 9 (2024). Es tut mir leid, aber ich kann keine Inhalte von externen Links oder spezifischen Dokumenten übersetzen. Wenn Sie den Text, den Sie übersetzt haben möchten, hier einfügen, helfe ich Ihnen gerne weiter.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen Methylierungsequenzierungsdiensten veröffentlicht wurden:
Genomisches Imprinting-ähnliches monoalleles väterliches Ausdrucksmuster bestimmt das Geschlecht von Kanalkatzenfischen.
Journal: Wissenschaftliche Fortschritte
Jahr: 2022
Folat-Träger-Defizienz fördert unterschiedliche Methylierung und erhöhte zelluläre Potenz an der neuralen Plattengrenze.
Zeitschrift: Frontiers in Cell and Developmental Biology
Jahr: 2022
Temporäres genomweites DNA-Methylierungssignal von post-smolt Pazifischen Lachsen, die mit Piscirickettsia salmonis herausgefordert wurden.
Journal: Epigenetik
Jahr: 2021
KMT2A assoziiert mit dem PHF5A-PHF14-HMG20A-RAI1 Subkomplex in Stammzellen des Pankreaskarzinoms und reguliert epigenetisch deren Eigenschaften.
Zeitschrift: Nature Communications
Jahr: 2023
Krebsassoziierte DNA-Hypermethylierung von Polycomb-Zielen erfordert die duale Erkennung von Histon H2AK119-Ubiquitinierung und der sauren Tasche des Nukleosoms durch DNMT3A.
Journal: Wissenschaftliche Fortschritte
Jahr: 2024
Genomisches Imprinting-ähnliches monoalleles väterliches Ausdrucksmuster bestimmt das Geschlecht von Kanalkatzenfischen.
Journal: Wissenschaftliche Fortschritte
Jahr: 2022
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


