
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Reduzierte Repräsentation Bisulfid-Sequenzierung
CD Genomics bietet einen Service für die reduzierte Repräsentations-Bisulfid-Sequenzierung (RRBS) mit Einzel-Nukleotid-Auflösung an, indem die benötigte Sequenzierungsmenge reduziert wird, während die Mehrheit der Promotoren und anderer relevanter genomischer Regionen erfasst wird. RRBS ist vorteilhaft für die Entdeckung von Biomarkern durch die Identifizierung und Analyse von unterschiedlich methylierten Regionen (DMRs) zwischen Proben.
Die Einführung von RRBS
Die DNA-Methylierung von Cytosin ist eine epigenetische Modifikation, die an der Regulierung der Genexpression beteiligt ist. Die DNA-Methylierung tritt überwiegend in CpG-Kontexten auf, und diese CpG-Dinukleotide sind in bestimmten Regionen des Genoms häufiger anzutreffen. RRBS ist ein leistungsstarker Ansatz zur genomweiten Analyse der DNA-Methylierung, der die Verdauung mit Restriktionsenzymen mit Bisulfit-Sequenzierung kombiniert, um einen CpG-dichten Anteil des Genoms anzureichern. Diese Kombination verbessert die Effizienz der Nutzung von Proben und bietet eine ideale Plattform für Pilotstudien und klinische Anwendungen.
RRBS sequenziert nur die Fragmente, die zu CpG-reichen Regionen gehören, zu deutlich reduzierten Kosten im Vergleich zu Whole-Genome-Bisulfid-Sequenzierung (WGBS)RRBS ist sehr kosteneffektiv, da etwa 1-5% des Genoms sequenziert werden, was etwa 12% der genomweiten CpG-Stellen und etwa 84% der CpG-Inseln in Promotoren abdeckt. Pflanzen zeigen eine andere CpG-Verteilung: (i) es fehlen charakteristische CpG-Inseln in Promotoren; (ii) sie treten an Cytosinbasen innerhalb aller Sequenzkontexte auf. Daher wird das Enzym MspI häufig bei Tieren und Menschen verwendet, während die Enzyme SacI/MseI oft bei Pflanzen eingesetzt werden.
Vorteile von RRBS
- Geringer DNA-Bedarf
- Methylierungsmuster von CpG-reichen Regionen im gesamten Genom
- Abdeckung von bis zu 5 Millionen CpG-Stellen im Menschen
- Gleichzeitige Erkennung von DNA-Methylierung und SNPs.
- Entdeckung epigenetischer Biomarker
- Einzel-Nukleotid-Auflösung und Kosten-Effizienz
Anwendungen von RRBS
Die reduzierte Repräsentation von Bisulfit-Sequenzierung kann für, aber nicht beschränkt auf, die folgenden Forschungsbereiche verwendet werden:
KrebsforschungDie Forschung zu Krebs nutzt die RRBS-Technologie, um DNA-Methylierungsprofile über verschiedene Krebsarten hinweg sorgfältig zu kartieren. Durch die Analyse der Unterschiede in den Methylierungsmustern zwischen krebsartigem und gesundem Gewebe hilft RRBS, krebs-spezifische Methylierungsmarker zu identifizieren. Dies beleuchtet die epigenetischen Prozesse, die an der Entstehung und Entwicklung von Krebs beteiligt sind. Darüber hinaus deckt RRBS spezifische Veränderungen in der DNA-Methylierung auf, die potenziell als frühe Indikatoren für innovative Diagnosestrategien genutzt werden könnten. Dieser Ansatz unterstreicht die entscheidende Rolle der Epigenetik in der Krebsforschung und betont die Suche nach neuartigen Biomarkern für eine frühzeitige Erkennung und Intervention.
EntwicklungsbiologieDie RRBS-Technologie ist ein wertvolles Werkzeug zur Untersuchung der vielschichtigen Veränderungen der DNA-Methylierung, die während der embryonalen Entwicklung auftreten. Sie ermöglicht eine umfassende Analyse von stadien- und zellspezifischen Methylierungsmustern und beleuchtet die komplexen Dynamiken der epigenetischen Regulation. Darüber hinaus erlaubt RRBS die Beobachtung von DNA-Methylierungsmodifikationen während der Differenzierung von Stammzellen und entschlüsselt die epigenetischen Mechanismen, die die Bestimmung des Zellschicksals und die Genexpression komplex orchestrieren.
NeurowissenschaftenIn der Neurowissenschaft untersucht RRBS die DNA-Methylierungsprofile bei neurologischen Erkrankungen wie Alzheimer und Autismus, enthüllt deren epigenetische Grundlagen und verbessert diagnostische sowie therapeutische Strategien. Darüber hinaus erforscht RRBS DNA-Methylierungsänderungen, die mit der Gedächtnisbildung und Lernprozessen verbunden sind, und fördert das Verständnis der epigenetischen Regulation in der neuronalen Funktion und im Verhalten.
Evolutions- und Ökologische StudienDie RRBS-Technologie dient als leistungsstarkes Werkzeug für vergleichende Studien zu DNA-Methylierungsmustern unter verschiedenen Arten oder Populationen im Bereich der evolutionären und ökologischen Forschung. Durch diese Methodik können Forscher die Auswirkungen der Epigenetik auf evolutionäre Dynamiken und adaptive Reaktionen untersuchen. Darüber hinaus beleuchtet RRBS die Auswirkungen von Umweltfaktoren wie Verschmutzung und Nährstoffvariationen auf die komplexe Modulation der DNA-Methylierung und deckt somit das komplexe Zusammenspiel zwischen epigenetischen Veränderungen und Umwelteinflüssen auf.
AgrarwissenschaftenIm Bereich der Agrarwissenschaften dient RRBS als entscheidendes Werkzeug zur Untersuchung von DNA-Methylierungsprofilen innerhalb von Pflanzensystemen, die für wichtige Merkmale wie Krankheitsresistenz, Ertragskapazität und Produktqualität relevant sind. Solche Analysen bieten entscheidende Hinweise für Zuchtinitiativen, die darauf abzielen, die Eigenschaften von Pflanzenvarianten zu verbessern. Darüber hinaus erkennt RRBS im Bereich der Tierzucht epigenetische Indikatoren, die mit begehrten Produktionsmerkmalen und dem allgemeinen Wohlbefinden verbunden sind, und liefert somit Erkenntnisse, die strategische Zuchtentscheidungen informieren und die Effizienz von Zuchtprogrammen steigern.
RRBS-Workflow
Der allgemeine Arbeitsablauf für RRBS-Sequenzierung ist unten skizziert. RRBS nutzt das Restriktionsenzym MspI, um an CCGG-Stellen im Genom zu schneiden und so CpG-dichte Regionen anzureichern. Die Regionen, die CpGs enthalten, werden dann durch Gelreinigung zurückgewonnen. Nach der Ligatur von methylierte Adaptern, Endreparatur und dA-Tailing werden diese Fragmente dann durch Bisulfit umgewandelt, amplifiziert und sequenziert. Unser hochqualifiziertes Expertenteam führt Qualitätsmanagement durch und überwacht jeden Schritt, um zuverlässige und unvoreingenommene Ergebnisse zu gewährleisten.

Dienstleistungsspezifikation
Musteranforderungen
|
|
 Klicken |
Sequenzierungsstrategien
|
 |
Datenanalyse Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an:
|
Analyse-Pipeline

Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details in reduzierter Darstellung der Bisulfit-Sequenzierung für Ihre Schreibweise (Anpassung)
Durch die Kombination von Enzymverdau, Bisulfitumwandlung und NGS-TechnologieCD Genomics kann RRBS als alternative Methode anbieten, um genomweite Methylome kostengünstiger zu erstellen und zu profilieren. Wenn Sie zusätzliche Anforderungen oder Fragen haben, zögern Sie bitte nicht, uns zu kontaktieren.
Referenzen:
- Guo H, Zhu P, Guo F, u. a.Profilierung der DNA-Methylom-Landschaften von Säugetierzellen mit Einzelzell-reduzierter Repräsentations-Bisulfid-Sequenzierung. Naturprotokolle, 2015, 10(5): 645.
- Meissner A, Gnirke A, Bell G W, u. a.Reduzierte Repräsentation Bisulfite-Sequenzierung für vergleichende hochauflösende DNA-Methylierungsanalyse. Nukleinsäureforschung, 2005, 33(18): 5868-5877.
Demonstrationsergebnisse
Teilweise Ergebnisse sind unten aufgeführt:

Häufig gestellte Fragen zur reduzierten Repräsentation Bisulfid-Sequenzierung
1. Was ist der Workflow für RRSB?
Der Arbeitsablauf von RRBS wird in Abbildung 1 unten dargestellt. Kurz gesagt, wird die DNA zunächst verdaut, typischerweise mit MspI, das methylierungsunempfindlich ist und an CCGG-Stellen schneidet, wodurch CpG-reiche Regionen des Genoms angereichert werden. Die Fragmentenden werden dann repariert und mit dA versehen, und Adapter werden ligiert. Anschließend werden die Fragmente nach Größe ausgewählt, durch Bisulfit umgewandelt, amplifiziert und sequenziert.
 Abbildung 1. Der schematische Arbeitsablauf für RRBS.
Abbildung 1. Der schematische Arbeitsablauf für RRBS.
2. Was sind die Anforderungen für die Sequenzierung von Proben?
Sie können Zellen einreichen (mindestens 5*10).6), frisches Gewebe (mindestens 2~3 g) oder DNA (mindestens 5 μg; Konzentration ≥ 100 ng/µl; OD = 1,8~2,0; ohne Degradation oder RNA-Kontamination oder schwere Degradation). Vor dem Versand sollten die Zellen bei -80°C gelagert werden, und die DNA sollte in Wasser gelöst und bei -20°C aufbewahrt werden. Bitte vermeiden Sie wiederholte Gefrier-Tau-Zyklen. Beim Versand sollten die Proben mit Parafilm versiegelt und wahrscheinlich mit Kühlakkus gefroren gehalten werden.
3. Welche Arten sind für die reduzierte Repräsentation von Bisulfit-Sequenzierung geeignet?
Die Arten, die RRBS unterzogen werden, sollten drei Anforderungen erfüllen: (i) Eukaryoten; (ii) ihr Referenzgenom sollte mindestens auf Scaffold-Ebene assembliert sein; (iii) relativ vollständige Genomanalysen.
Reduzierte Repräsentation Bisulfite-Sequenzierung Fallstudien
DNA-Methylierungsänderungen, die durch lange und kurze Photoperioden induziert werden in Nasonia
Journal: Genomforschung
Impact-Faktor: 11,922
Veröffentlicht: 15. Dezember 2015
Zusammenfassung
Wespe Nasonia vitripennis ist ein aufkommendes Modellorganismus, das eine starke photoperiodische Reaktion zeigt und verwendet werden kann, um die molekularen Mechanismen zu untersuchen, die der photoperiodischen Zeitmessung zugrunde liegen. Die Autoren versuchten herauszufinden, wie Weibchen Nasonia die Entwicklungsbahn ihrer Nachkommen zu steuern, um den Winter zu überstehen, indem sie die RRBS-Methode nutzen. Es wurde gezeigt, dass das Herunterregulieren von DNA-Methyltransferasen oder das Blockieren der DNA-Methylierung die photoperiodische Diapause-Reaktion der Wespen erheblich stört, was auf die Rolle von hinweist. DNA-Methylierung in der photoperiodischen Zeitmessung von Insekten.
Materialien & Methoden
- Wespenstämme
- Wildtyp-Stamm, AsymC
- genomische DNA-Extraktion
- Bibliothekskonstruktion
- RRBS-Sequenzierung
- Illumina HiSeq 2000 Analysator
- hmeDIP Protokoll
- Qualitätskontrolle
- Lesefilterung und Ausrichtung
- Differenzielle Methylierungsanalyse
- Funktionale Tests
Ergebnisse
1. Der Nasonia Methylom
Die Autoren profilierten die DNA-Methylierung in genomischer DNA, die aus weiblichen Proben extrahiert wurde. N. vitripennis unter Lang- oder Kurztagbedingungen gehalten, indem RRBS genutzt wurde. Die CpG-Methylierung erklärt einen großen Teil der Überlappung zwischen den beiden Proben im Gegensatz zu anderen Sequenzkontexten (Abbildung 1B), was darauf hindeuten könnte, dass die Nicht-CpG-Methylierung größtenteils experimentelles Rauschen darstellt. CpGs scheinen entweder stark oder schwach methyliert zu sein (Abbildung 1C). Die Mehrheit der methylierten CpGs befand sich in Exons, und die Mehrheit war in Introns, Promotorregionen und intergenen Regionen lokalisiert (Abbildung 1D, E).
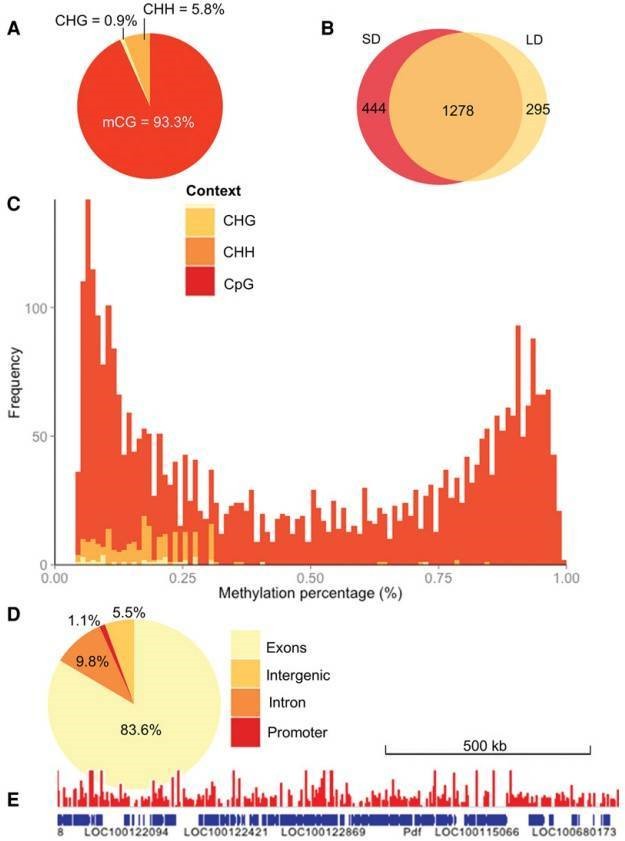 Abbildung 1. Die Nasonia Methylom.
Abbildung 1. Die Nasonia Methylom.
2. Identifizierung von differentiell methylierten Genen
Die Autoren identifizierten 51 unterschiedlich methylierte CpG-Stellen (DMCs), die auf 37 Gene kartiert wurden. Etwa die Hälfte der DMCs (23/51) zeigte eine Hypomethylierung bei langen Tagen im Vergleich zu kurzen Tagen, während die anderen den entgegengesetzten Trend aufwiesen. qPCRs wurden durchgeführt, um die RRBS-Analyse zu validieren.
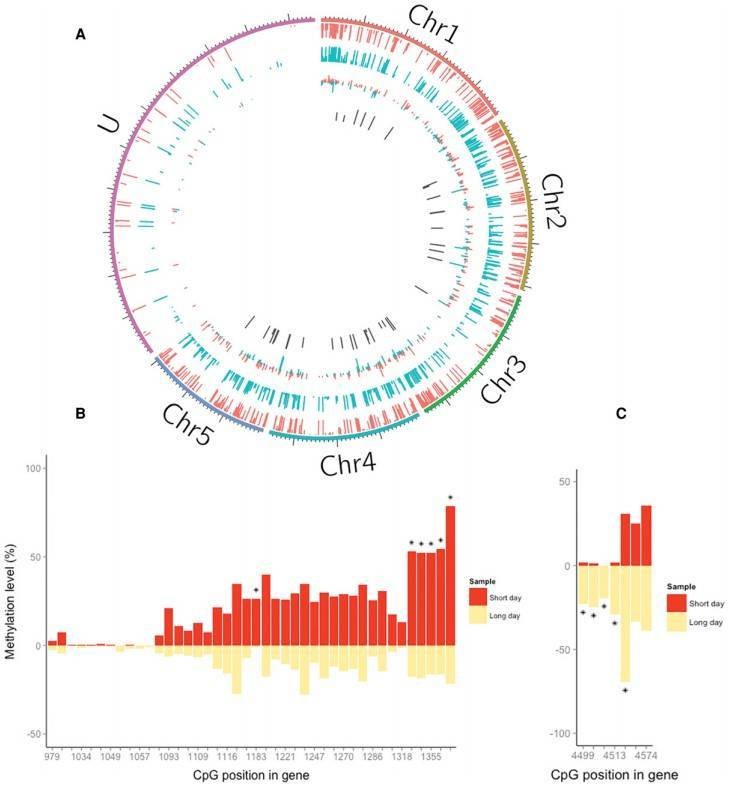 Abbildung 2. Differenzielle DNA-Methylierung im Zusammenhang mit Photoperioden in Nasonia.
Abbildung 2. Differenzielle DNA-Methylierung im Zusammenhang mit Photoperioden in Nasonia.
3. Funktionelle Assays, die eine ursächliche Rolle der DNA-Methylierung nachweisen
Die Autoren schalteten Dnmt1a-c und Dnmt3 aus, indem sie dsRNA in weibliche Puppen der N. vitripennis Stamm AsymC. Der Knockdown wurde durch qPCR verifiziert. Der Knockdown von Dnmt1a führte dazu, dass Weibchen unabhängig von der Tageslänge Diapause-Nachkommen produzierten. Das Stummschalten von Dnmt3 ergab einen nicht signifikanten (aber nahe an der Signifikanz) Unterschied in der Diapause-Reaktion, was darauf hindeutet, dass der Effekt von Dnmt3 viel schwächer oder schwerer zu erfassen ist.
Sie testeten auch den Einfluss der DNA-Methylierung pharmakologisch, indem sie den DNA-Methylierungshemmer 5-Aza-2'-deoxy-cytidin (5-aza-dC) verwendeten, und beobachteten eine Verringerung der Diapause bei den Nachkommen von Wespen, die in kurzen Tagen gehalten wurden, sowie eine Zunahme bei den Nachkommen in langen Tagen.
 Abbildung 3. DNA-Methylierung ist erforderlich für die photoperiodisch vermittelte Diapause-Reaktion.
Abbildung 3. DNA-Methylierung ist erforderlich für die photoperiodisch vermittelte Diapause-Reaktion.
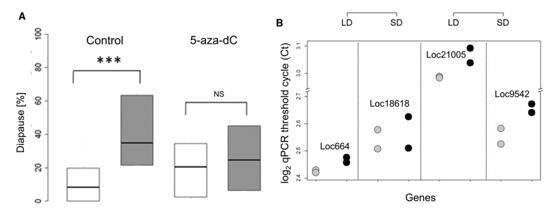 Abbildung 4. Pharmakologische Tests mit 5-aza-dC.
Abbildung 4. Pharmakologische Tests mit 5-aza-dC.
Fazit
Die Autoren fanden heraus, dass die DNA-Methylierung in Nasonia vitripennis Änderungen mit dem saisonalen Photoperiod. Die Störung wichtiger Methylierungsenzyme (Dnmt1a oder die Verwendung von 5-aza-dC) verändert, wie Weibchen auf Photoperioden reagieren. Ihre hochauflösenden Methylomdaten zeigen, dass die Methylierungsmuster von Nasonia genkörperzentriert sind, ähnlich wie bei anderen Insekten. Dies bestätigt die Rolle der DNA-Methylierung bei der Photoperiodenreaktion und deutet darauf hin, dass verschiedene Methyltransferasen unterschiedliche Funktionen in diesem Prozess haben.
Referenz:
-
Pegoraro M, Bafna A, Davies N J, u. a.DNA-Methylierungsänderungen, die durch lange und kurze Photoperioden induziert werden in Nasonia. Genomforschung, 2016, 26(2): 203-210.
Verwandte Veröffentlichungen
Hier sind einige Veröffentlichungen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Folat-Träger-Defizienz fördert unterschiedliche Methylierung und erhöhte zelluläre Potenz an der neuralen Plattenrand.
Zeitschrift: Frontiers in Cell and Developmental Biology
Jahr: 2022
Temporäres genomweites DNA-Methylierungssignal von post-smolt Pazifischen Lachsen, die mit Piscirickettsia salmonis herausgefordert wurden.
Journal: Epigenetik
Jahr: 2021
Fettreiche Diäten während der Schwangerschaft führen zu Veränderungen der DNA-Methylierung und Proteinexpression im Pankreasgewebe der Nachkommen: Ein Multi-Omics-Ansatz
Zeitschrift: Internationale Zeitschrift für Molekulare Wissenschaften
Jahr: 2024
Die Analyse der reduzierten Repräsentations-Bisulfid-Sequenzierung (RRBS) zeigt Variationen in der Verteilung und den Levels der DNA-Methylierung in der weißen Birke (Betula papyrifera), die Nickel ausgesetzt ist.
Journal: Genom
Jahr: 2024
Krebsassoziierte DNA-Hypermethylierung von Polycomb-Zielen erfordert die doppelte Erkennung von Histon H2AK119-Ubiquitinierung und der sauren Tasche des Nucleosoms durch DNMT3A.
Journal: Wissenschaftliche Fortschritte
Jahr: 2024
Egr2-Deletion in autoimmunen anfälligen C57BL6/lpr-Mäusen unterdrückt die Expression von methylierungsempfindlichen Dlk1-Dio3-Cluster-Mikro-RNAs.
Journal: ImmunoHorizons
Jahr: 2023
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


