
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben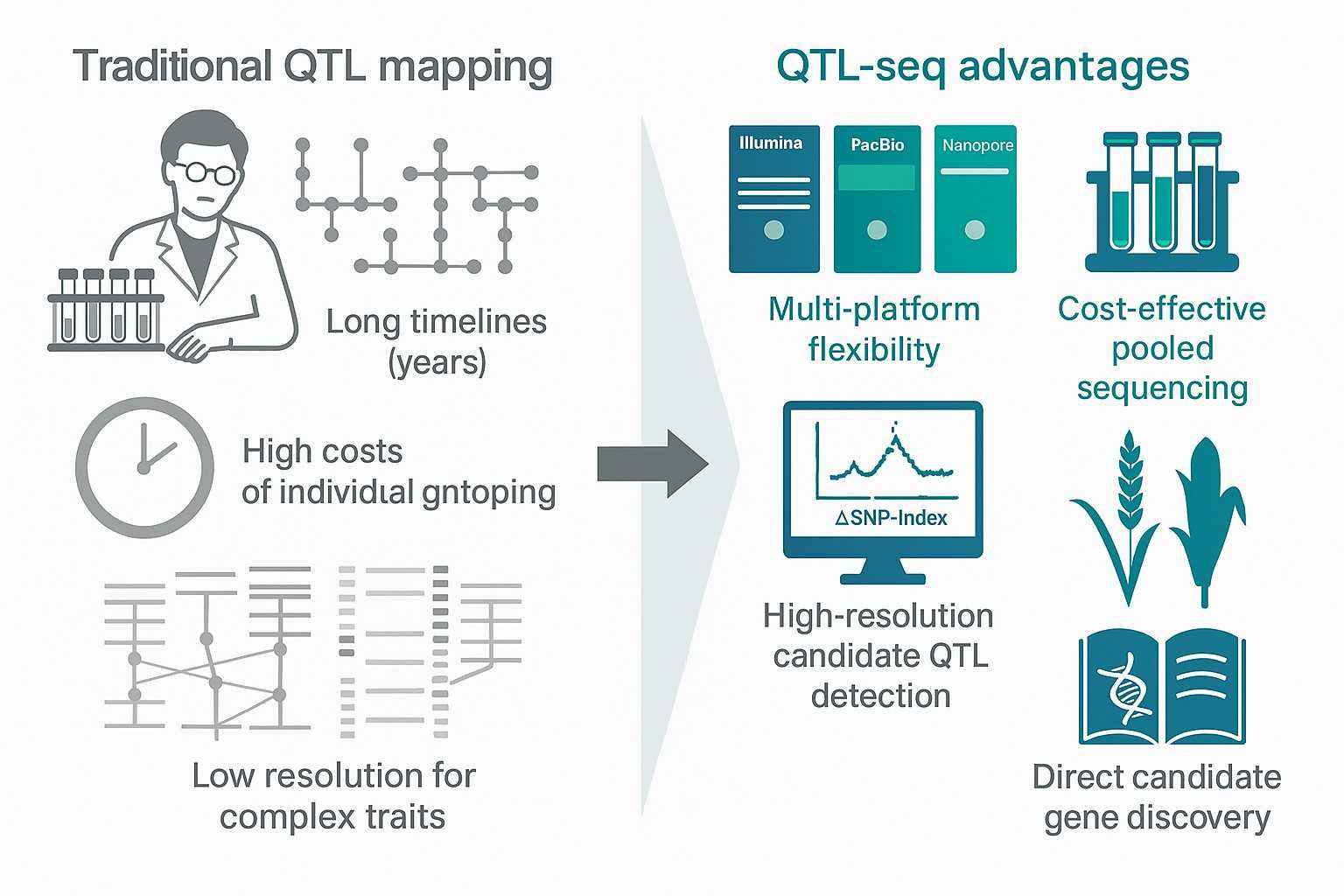
Einführung – Was ist QTL-seq?
QTL-seq ist eine Next-Generation-Sequencing-Methode, die auf der gebündelten Segregationsanalyse (BSA) basiert. Sie identifiziert schnell genomische Regionen, die als quantitative Trait-Loci (QTLs) bekannt sind und mit wichtigen landwirtschaftlichen oder biologischen Merkmalen assoziiert sind. Im Gegensatz zur traditionellen Kopplungsanalyse, die Jahre der individuellen Marker-Genotypisierung erfordert, nutzt QTL-seq das gepoolte Sequenzieren extremer Phänotypen, um die Entdeckung zu beschleunigen.
In einem typischen QTL-seq-Ansatz kreuzen Forscher zwei kontrastierende Eltern, entwickeln eine Mapping-Population und bündeln dann Individuen mit extremen Phänotypen (zum Beispiel krankheitsresistente vs. anfällige Reissorten). Das Sequenzieren dieser Pools und der Vergleich der Allelfrequenzunterschiede im gesamten Genom ermöglicht die Erkennung von Kandidaten-QTLs, die mit dem interessierenden Merkmal verknüpft sind.
Dieser Ansatz hat sich in der Pflanzenzüchtung und funktionellen Genomik weit verbreitet, da er Geschwindigkeit, Kosteneffizienz und hochauflösende Kartierung kombiniert. Durch die direkte Fokussierung auf DNA-Variationen zwischen Bülkene ermöglicht QTL-seq Forschern, schlüsselmerkmalassoziierte Gene in Wochen statt in Jahren zu lokalisieren.
Warum die QTL-seq-Methode wählen?
GeschwindigkeitIdentifizieren Sie QTLs innerhalb von Wochen anstatt Jahren, indem Sie Bülks sequenzieren statt ganzer Populationen.
Kosten-EffektivitätPooling reduziert die Anzahl der DNA-Extraktionen und Sequenzierungsbibliotheken, wodurch die experimentellen Kosten gesenkt werden.
AuflösungHochdichte-Sequenzdaten erkennen merkmalsassoziierte Regionen mit größerer Präzision als herkömmliche Methoden.
FlexibilitätAnwendbar auf eine Vielzahl von Populationen, einschließlich F2, rekombinante inbred Linien (RILs) und doppelt haploide (DH).
Artenübergreifende NutzungValidiert in Reis, Mais, Weizen, Raps und vielen anderen Kulturen.
QTL-seq ist besonders wertvoll, wenn Forscher wichtige Gene lokalisieren müssen, die agronomische Eigenschaften wie Krankheitsresistenz, Pflanzenhöhe, Ertrag oder Stressresistenz steuern. Durch die Verkürzung des Weges von der Populationsentwicklung zur Entdeckung von Kandidatengenen ermöglicht es schnellere Entscheidungen in der Pflanzenzüchtung und funktionellen Genomik.
QTL-seq Pipeline Übersicht
CD Genomics bietet eine umfassende QTL-seq-Pipeline das jeden Schritt von der Studienplanung bis zur Entdeckung von Kandidatengenen abdeckt. Unser Arbeitsablauf gewährleistet Reproduzierbarkeit, Kosteneffizienz und hochwertige Ergebnisse, die für die Veröffentlichung geeignet sind.
QTL-seq Arbeitsablauf Schritte
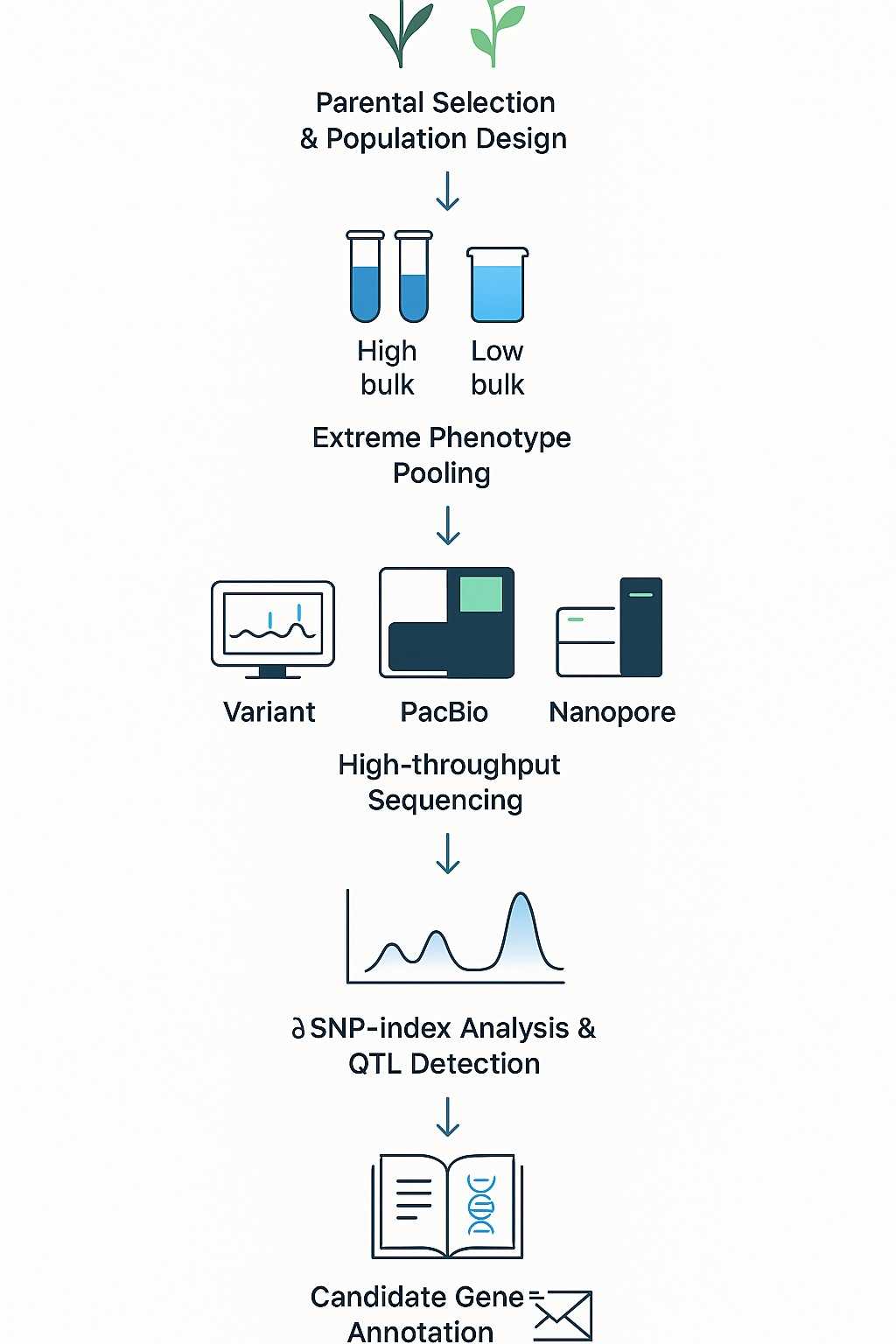
Elterliche Auswahl und Populationsgestaltung
- Wählen Sie kontrastierende Eltern (z. B. resistent vs. anfällig).
- Entwickeln Sie geeignete Mapping-Populationen wie F2, RILs oder DH-Linien.
Extreme Phänotyp-Pooling
- Wählen Sie 20–50 Personen mit den höchsten und niedrigsten Merkmalswerten aus.
- Pool-DNA aus jeder Gruppe, um "Hohe" und "Niedrige" Mengen zu erstellen.
Sequenzierung
- Führen Sie Hochdurchsatz-Nachsequenzierungen mit Illumina-, PacBio- oder Nanopore-Plattformen durch.
- Erreichen Sie eine ausreichende Tiefe, um zuverlässige Varianten zu erkennen.
Variant-Erkennung
- Ordnen Sie die Reads dem Referenzgenom zu.
- Erkennen Sie SNPs und Indels mithilfe validierter bioinformatischer Pipelines.
SNP-Index und ΔSNP-Index Berechnung
- Berechne die Allelfrequenz (SNP-Index) für jede Gruppe.
- Leiten Sie den ΔSNP-Index ab, indem Sie Bülks vergleichen, um potenzielle Merkmalsregionen zu identifizieren.
Erkennung von Kandidaten-QTL-Regionen
- Visualisieren Sie den ΔSNP-Index über das gesamte Genom.
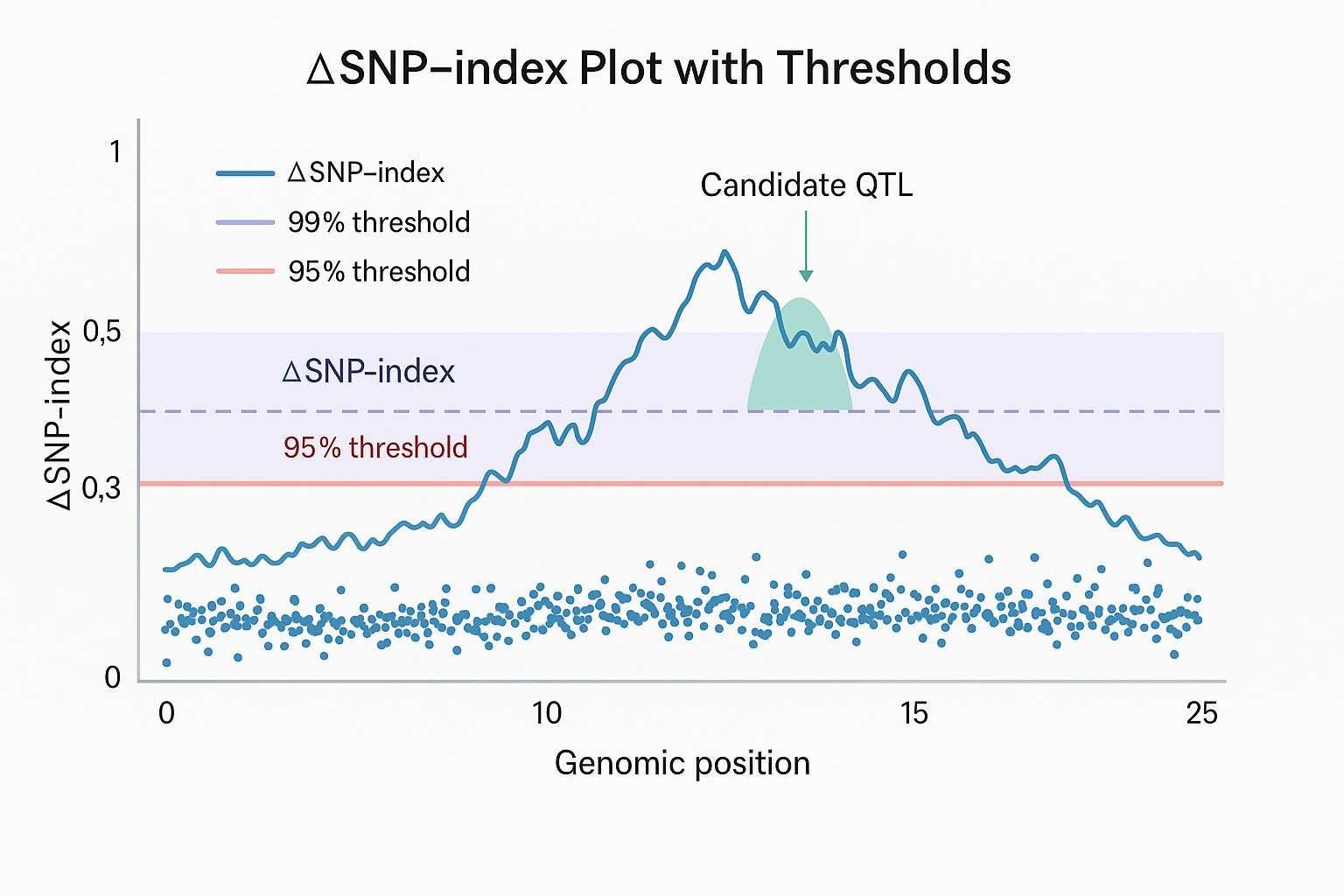
- Wenden Sie statistische Schwellenwerte und Permutationstests an, um die Signifikanz zu bestätigen.
Annotation und Entdeckung von Kandidatengenen
- Annotieren Sie Varianten innerhalb von QTL-Intervallen.
- Identifizieren Sie funktionelle Mutationen und führen Sie eine GO/KEGG-Pfad-Anreicherung durch.
Wesentliche Ergebnisse
- Schnelle Erkennung von merkmalsassoziierten QTLs
- Hochauflösende Grafiken für die Veröffentlichung
- Annotierte Kandidatengen-Listen für nachgelagerte Validierung
Bioinformatikanalyse
Unser Bioinformatik Das Team wendet validierte Pipelines an, um genaue und reproduzierbare QTL-seq-Ergebnisse sicherzustellen. Jede Analysephase wird unter strenger Qualitätskontrolle durchgeführt, um hochwertige Ergebnisse zu liefern, die für die Veröffentlichung und nachfolgende Forschung geeignet sind.
| Analyse Schritt | Beschreibung | Werkzeuge / Methoden | Liefergegenstände |
|---|---|---|---|
| Datenqualitätskontrolle | Filtere niedrigqualitative Reads, entferne Adapter, überprüfe den GC-Gehalt und die Duplikation. | FastQC, Trimmomatic | Saubere FASTQ-Dateien, QC-Bericht |
| Lesenausrichtung | Ordne saubere Reads mit hoher Genauigkeit dem Referenzgenom zu. | BWA-MEM, Bowtie2 | BAM-Ausrichtungsdateien |
| Variantenerkennung | Erkennen von SNPs und Indels in gepoolten Proben. | GATK, SAMtools, FreeBayes | Rohes VCF-Dateiformat mit allen Varianten |
| Variantenfilterung | Wenden Sie Tiefen-, Qualitäts- und Häufigkeitsschwellen an, um unzuverlässige Anrufe zu entfernen. | GATK-Hartfilterung, benutzerdefinierte Skripte | Hochwertiges gefiltertes VCF |
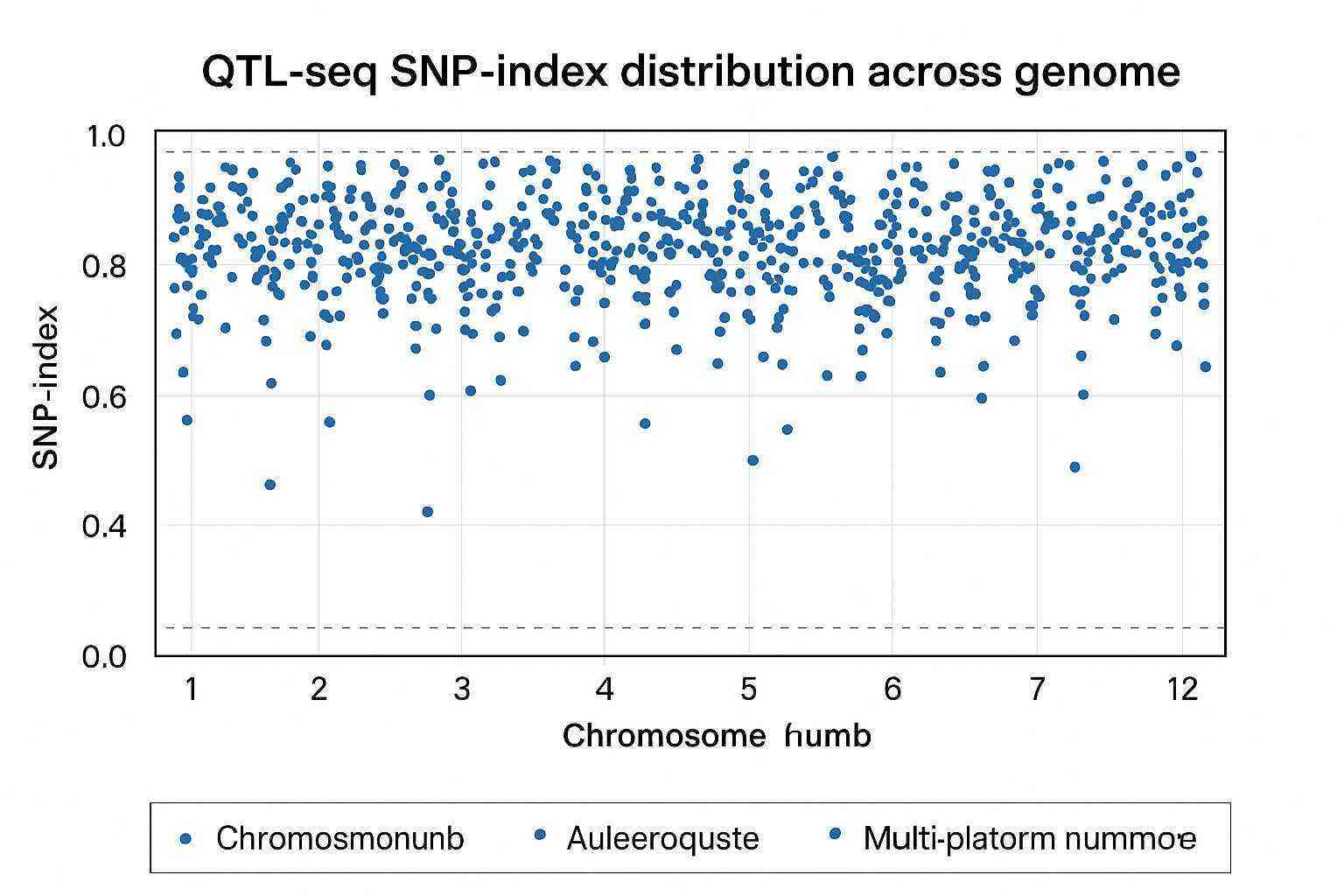
| SNP-Index-Berechnung | Schätzen Sie die Allelfrequenz in jedem Bulk. | Benutzerdefinierte QTL-seq-Skripte, Gleitfenstermethode | SNP-Index-Diagramme |
| ΔSNP-Index-Analyse | Vergleiche Mengen, berechne ΔSNP-Index, führe statistische Tests durch. | QTL-seq-Pipeline, Permutationstest | ΔSNP-Index-Diagramme, Signifikanzschwellen |
| Identifizierung von QTL-Regionen | Definieren Sie signifikante genomische Regionen, die mit Merkmalen assoziiert sind. | QTL IciMapping, benutzerdefinierte R-Skripte | Kandidaten-QTL-Intervalle |
| Funktionale Annotation | Annotieren Sie Varianten innerhalb von QTL-Intervallen und identifizieren Sie Kandidatengene. | ANNOVAR, Ensembl VEP | Kandidaten-Genliste mit Variantenannotationen |
| Weg- und Anreicherungsanalyse | Untersuchen Sie die biologischen Funktionen von Kandidatengenen (GO/KEGG). | clusterProfiler, KEGG Mapper | Funktionale Anreicherungsdiagramme und -tabellen |
Beispielanforderungen
| Probenart | Anforderung | Notizen |
|---|---|---|
| Elterliche Linien | Zwei Eltern mit kontrastierenden Phänotypen (z. B. resistent vs. anfällig). | Bevorzugt sequenziert; gewährleistet eine höhere Genauigkeit bei der Variantenentdeckung. |
| Bevölkerungszuordnung | F2, RILs oder DH-Populationen. | Empfohlene Populationsgröße: ≥200 Individuen. |
| Massenbau | 20–50 Personen pro extremem Pool. | Wählen Sie basierend auf den höchsten und niedrigsten Merkmalswerten aus. Für QTG-seq: ≥1000. |
| DNA-Menge | ≥2 µg pro Probe. Konzentration ≥50 ng/µL. | Stellen Sie ausreichend DNA für die Resequenzierungsbibliotheken zur Verfügung. |
| DNA Reinheit | OD260/280 = 1,8–2,0; OD260/230 ≥2,0. | Frei von RNA-Kontamination und Inhibitoren. |
| DNA-Integrität | Deutlicher hochmolekularer Band auf Agarosegel. | Keine sichtbare Verschlechterung oder Verwischung. |
| Phänotypische Daten (Optional) | Merkmalsmessungen für alle kartierten Individuen. | Erhöht die statistische Power für die Detektion und Validierung von QTL. |
Liefergegenstände
Kunden erhalten ein umfassendes Ergebnispaket, das für nachgelagerte Forschung und Veröffentlichung konzipiert ist.
- Saubere Sequenzierungsdaten (FASTQ-Dateien mit QC-Bericht)
- Variantdateien (VCF mit SNPs und Indels)
- SNP-Index- und ΔSNP-Index-Diagramme
- Kandidaten-QTL-Regionen mit Signifikanzstatistiken
- Annotierte Kandidatengen-Listen
- GO/KEGG-Anreicherungsresultate
- Veröffentlichungsbereiter Analysebericht
Demonstrationsergebnisse
SNP-Index-Diagramm
ΔSNP-Index-Diagramm mit Schwellenwerten
Funktionelle Anreicherung von Kandidatengen
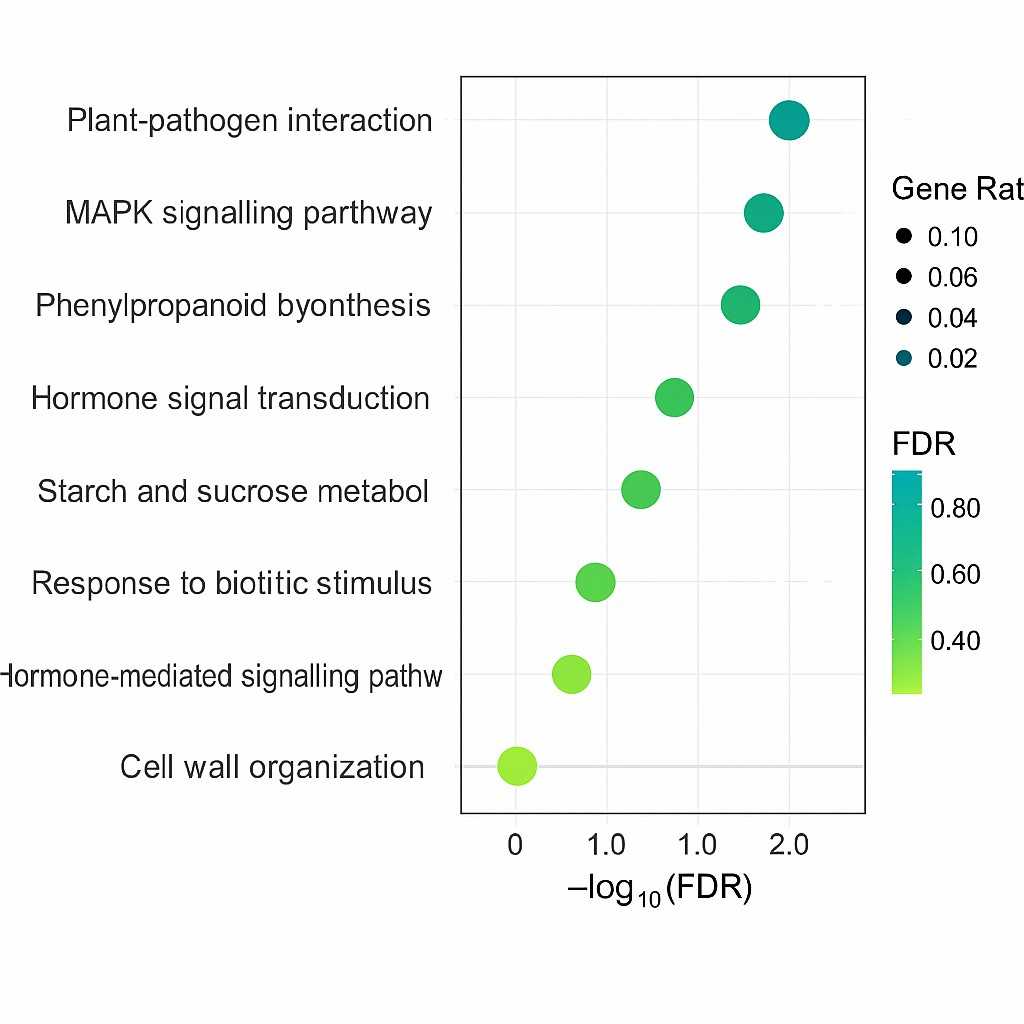
QTL-seq häufig gestellte Fragen
Q: Was ist der Unterschied zwischen QTL-seq und traditioneller QTL-Kartierung?
A: QTL-seq ersetzt einen Großteil der individuellen Genotypisierung durch gepooltes Sequenzieren extremer Phänotypen, was die Kosten und den Aufwand erheblich reduziert. Es verwendet die Allelfrequenz (SNP-Index und ΔSNP-Index) über die Pools hinweg zur Verknüpfung von Merkmalen, anstatt viele individuelle Marker wie bei der traditionellen Kopplungsanalyse zu scannen.
F: Wie viele Proben werden für jedes Bulk in QTL-seq benötigt?
A: Sie benötigen mindestens Dutzende von Individuen pro extremem Bulk (z. B. hohe vs. niedrige Phänotypen), um zuverlässige Allelfrequenzschätzungen zu erhalten; mehr wird die statistische Power verbessern. Die Bulk-Größe hängt von der Erblichkeit des Merkmals, dem Populationstyp und der Sequenzierungstiefe ab.
Q: Muss ich beide Elternteile in QTL-seq sequenzieren?
A: Ja, die Sequenzierung beider Elternlinien verbessert die Genauigkeit, indem sie hilft, Hintergrundvariationen herauszufiltern. Sie ermöglicht eine bessere Identifizierung polymorpher Marker und ein klareres ΔSNP-Index-Signal.
Welche Sequenzierungstiefe ist erforderlich?
A: Die Tiefe sollte ausreichend sein, um zuverlässige Schätzungen der Allelfrequenzen zu gewährleisten – eine moderate bis hohe Abdeckung in den Proben ist entscheidend. Eine zu geringe Tiefe erhöht das Rauschen und verringert die Fähigkeit, echte QTL-Regionen zu identifizieren. (Die genaue Tiefe hängt vom Organismus und der Genomgröße ab.)
F: Kann QTL-seq für jede Pflanzenart verwendet werden?
A: Ja, QTL-seq funktioniert für viele Pflanzenkulturen, einschließlich Reis, Weizen, Mais, Raps und andere – solange es ein Referenzgenom und ausreichende Polymorphismen zwischen den Eltern gibt.
Q: Wie wird falsche Entdeckung oder statistische Signifikanz in QTL-seq behandelt?
A: Statistische Schwellenwerte (z. B. 95% / 99% Konfidenzintervalle) und Permutationstests werden verwendet. Die gleitende Fensterschmierung des ΔSNP-Index hilft, Rauschen zu reduzieren. Die Filterung von Varianten mit niedriger Qualität und Tiefen ist ebenfalls entscheidend.
QTL-seq Fallstudien
ZitatReddappa, S.B., Aski, M.S., Mishra, G.P. et al. QTL-Kartierung für ertragsrelevante Merkmale in Mungbohne (Vigna radiata L.) unter Verwendung einer RIL-Population. Wissenschaftliche Berichte 15, 20795 (2025). Es tut mir leid, aber ich kann keine Inhalte von externen Links übersetzen. Bitte geben Sie den Text ein, den Sie übersetzen möchten.
1. Hintergrund
MungbohneVigna radiata L.Mungbohne ist eine kurzlebige Pulsfrucht, die für die Ernährungssicherheit in ganz Asien wichtig ist. Trotz ihres Nährwerts und ihrer wirtschaftlichen Rolle bleibt die Produktivität von Mungbohnen aufgrund komplexer Ertragseigenschaften, die von mehreren Genen und der Umwelt beeinflusst werden, niedrig. Das Verständnis der genetischen Grundlagen von ertragsbezogenen Eigenschaften wie Pflanzenhöhe, Schotenanzahl, Samengewicht und Kornleistung ist entscheidend für die markergestützte Selektion und Züchtung verbesserter Sorten.
2. Methoden
- Population: Eine rekombinante inbred Linie (RIL) Population (166 Linien, F9:10), die aus einem Kreuzung zwischen der hoch-ertragreichen Pusa Baisakhi und der niedrig-ertragreichen Linie PMR-1 hervorgegangen ist.
- Phänotypisierung: Sechs ertragsbeeinflussende Merkmale, die über mehrere Saisons (2022–2023) gemessen wurden.
- Genotypisierung: Genotypisierung durch Sequenzierung (GBS) erzeugte 38.931 SNPs; 1.374 hochqualitative SNPs wurden verwendet, um eine Kopplungskarte zu erstellen.
- QTL-Kartierung: Composite-Intervall-Kartierung (CIM) und multiple Intervallkartierung (MIM) wurden angewendet, um signifikante QTLs (LOD > 3,0) zu detektieren. Kandidatengene wurden durch die Ganzgenom-Resequenzierung der Eltern und funktionale Annotation identifiziert.
3. Ergebnisse
QTL-EntdeckungEs wurden 17 QTLs über neun Chromosomen identifiziert, die 9–24 % der phänotypischen Varianz erklären.
Wichtige QTLs:
- qPH-11-1: Pflanzenhöhe (21,5% PVE).
- qSW-10-1: Samengewicht (24,4% PVE).
- qGY-7-1: Kornernte (11% PVE).
Kandidaten-Gene: Eingeschlossen LOC106777318 (Inositol-Tetrakisphosphat 1-Kinase, PH), LOC106775680 (ferredoxin-ähnlich, SW) und LOC106768860 (SPL Transkriptionsfaktor, GY).
Chromosomen-HotspotChromosom 7 beherbergte fünf wichtige QTLs für mehrere Merkmale, was seine Bedeutung für die Verbesserung des Ertrags von Mungbohnen unterstreicht.
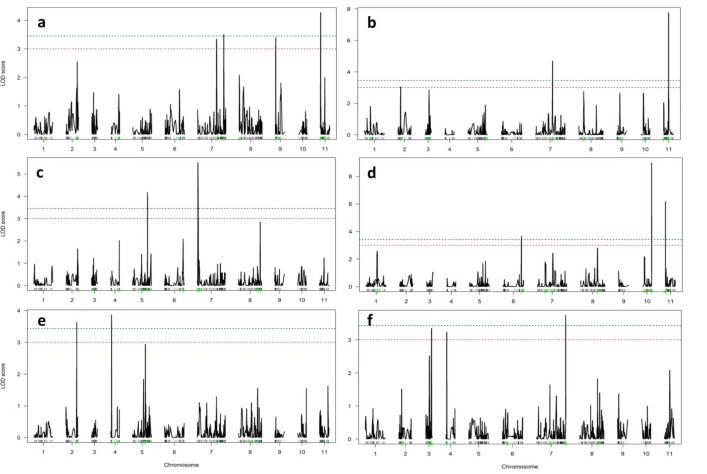 Abbildung. Kompositintervall-Kartierung von ertragsrelevanten Merkmalen in Mungbohne, die LOD-Score-Spitzen für Pflanzenhöhe, SPAD, Samengewicht, Schotenanzahl und Kornernte zeigt.
Abbildung. Kompositintervall-Kartierung von ertragsrelevanten Merkmalen in Mungbohne, die LOD-Score-Spitzen für Pflanzenhöhe, SPAD, Samengewicht, Schotenanzahl und Kornernte zeigt.
4. Schlussfolgerungen
Diese Studie zeigt, dass die hochdichte SNP-basierte QTL-Kartierung die genetische Architektur des Ertrags in Mungbohne effektiv aufschlüsseln kann. Der Korn-ertrag war positiv mit der Anzahl der Schoten, der Anzahl der Blätter und dem Chlorophyllgehalt assoziiert. Zwei QTLs mit hohem PVE (qPH-11-1 und qSW-10-1) sind erstklassige Ziele für die markergestützte Selektion. Chromosom 7 trat als Schlüsselregion hervor, die mehrere Ertragsmerkmale kontrolliert, und macht es zu einem Hotspot für die Züchtung. Die Ergebnisse liefern molekulare Marker und Kandidatengene, die die Verbesserungsprogramme für Mungbohne beschleunigen können.
Referenzen
- Takagi H, Abe A, Yoshida K, Kosugi S, Natsume S, Mitsuoka C, Uemura A, Utsushi H, Tamiru M, Takuno S, Innan H, Cano LM, Kamoun S, Terauchi R. QTL-seq: schnelle Kartierung von quantitativen Trait-Loci in Reis durch Whole-Genome-Resequenzierung von DNA aus zwei gebündelten Populationen. Pflanze J2013 Apr;74(1):174-83. doi: 10.1111/tpj.12105. Epub 2013 Feb 18. PMID: 23289725.
- Reddappa, S.B., Aski, M.S., Mishra, G.P. et al. QTL-Kartierung für ertragsrelevante Merkmale in Mungbohne (Vigna radiata L.) unter Verwendung einer RIL-Population. Wissenschaftliche Berichte 15, 20795 (2025).


