
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Gesamt-RNA-Sequenzierung
Die Total RNA-Seq-Analysen bieten einen umfassenden Überblick über das Transkriptom. Eine Reihe von Lösungen, die auf Ihre Studienziele zugeschnitten sind, ist bei CD Genomics verfügbar. Wir können einen schnellen, zuverlässigen und kosteneffektiven Service anbieten.
Die Einführung der Total-RNA-Sequenzierung
Da nicht-kodierende RNA (ncRNA) weiterhin als biologisch wichtig anerkannt wird, analysiert die totale RNA-Seq sowohl kodierende als auch mehrere Formen von nicht-kodierender RNA für einen umfassenden Überblick über das Transkriptom. Die globale Analyse der RNA-Expression kann unser Verständnis aller transkriptionellen Aktivitäten verbessern. Die totale RNA-Seq ermöglicht die Analyse von kodierender und nicht-kodierender RNA mit der hochzuverlässigen Entdeckung von Merkmalen wie alternativen Transkripten, Genfusionen, allelspezifischer Expression und der Erkennung neuartiger Transkripte in sowohl kodierenden als auch nicht-kodierenden RNA-Arten.
CD-Genomik kombiniert bewährte Chemien zur Vorbereitung von Ribosomenreduktionsbibliotheken mit Illumina. NGS Technologie in ein einheitliches, optimiertes Protokoll. Die Ribo-Zero-Ribosomen-RNA-Reduktionschemie minimiert die ribosomale Kontamination und maximiert den Prozentsatz der einzigartig zugeordneten Reads. Mit der konstanten robusten Leistung deckt das totale RNA-seq sowohl ab mRNA und eine breite Palette von ncRNA-Spezies von Interesse, einschließlich langes nicht-kodierendes RNA (lncRNA)kleine nukleäre RNA (snRNA), kleine nucleoläre RNA (snoRNA) und andere RNA-Spezies.
Vorteile der Total-RNA-Sequenzierung
- Präzise Messung der Faserorientierung und gleichmäßige Abdeckung.
- Kompatibel mit mehreren Probenarten, einschließlich niedrigqualitativer, formalinfixierter, paraffin-eingebetteter (FFPE) Proben.
- Schnelle Bearbeitungszeit und höchste Datenqualität
- Niedrigere Kosten und breite Verfügbarkeit.
- Effektive Transkriptomanalyse
- Die Forschung zur Transkriptom-Sequenzierung, die Genfusionen, Indels, SNPs und andere umfasst, führt zu höheren Erkennungsraten und verbesserter Zuverlässigkeit.
- Entdeckung neuer Transkripte und Spleißvarianten.
- Allelspezifische Genexpressionsanalyse.
Gesamt-RNA-Sequenzierungs-Workflow
Unsere umfassenden RNA-Seq-Dienste bieten den RNA-Sequenzierungsworkflow von der Probenvorbereitung bis zur Datenanalyse und ermöglichen eine schnelle Profilierung sowie tiefgehende Einblicke in die RNA.

Dienstspezifikationen
Beispielanforderungen
|
|
 Klicken |
Sequenzierungsstrategie
|
 |
Bioinformatikanalyse Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an:
|
Analyse-Pipeline
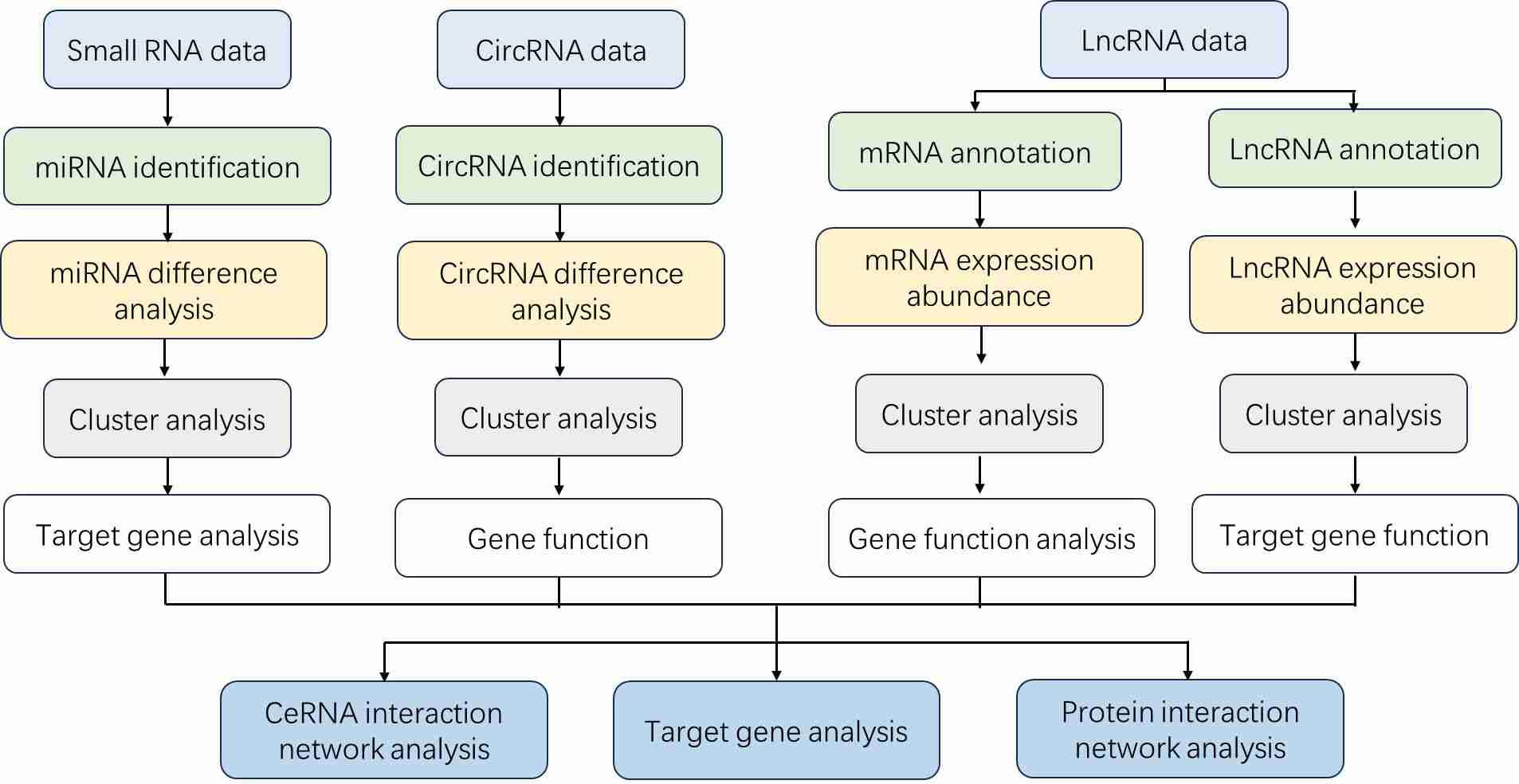
Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details zur Total-RNA-Sequenzierung für Ihre Schreibanpassung.
Unterstützt von unserem Team erfahrener Wissenschaftler und fortschrittlicher Technologie bietet CD Genomics Total RNA-Sequenzierungsdienste an. Wir wenden strenge Qualitätskontrollmaßnahmen und fortschrittliche bioinformatische Analysen an, um genaue und umfassende Ergebnisse zu gewährleisten. Wenn Sie spezifische Anforderungen oder Fragen haben, zögern Sie bitte nicht, uns für weitere Unterstützung zu kontaktieren.
Demo-Ergebnisse
Teilweise Ergebnisse sind unten aufgeführt:

Sequenzierungsqualitätsverteilung
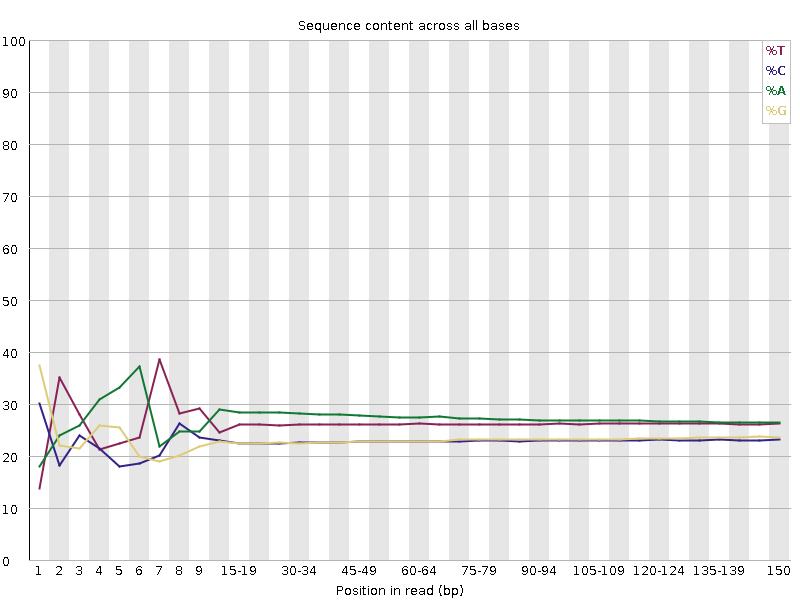
A/T/G/C-Verteilung
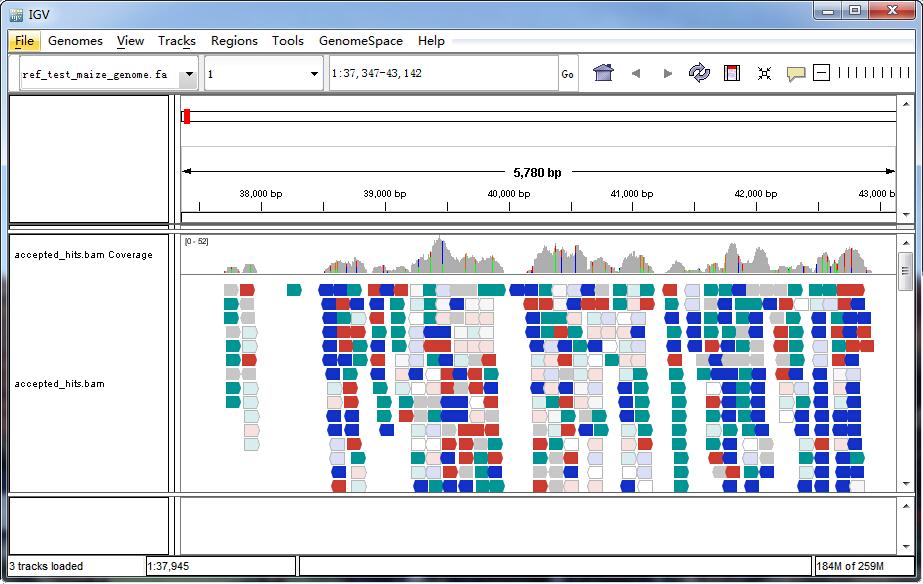
IGV-Browser-Oberfläche
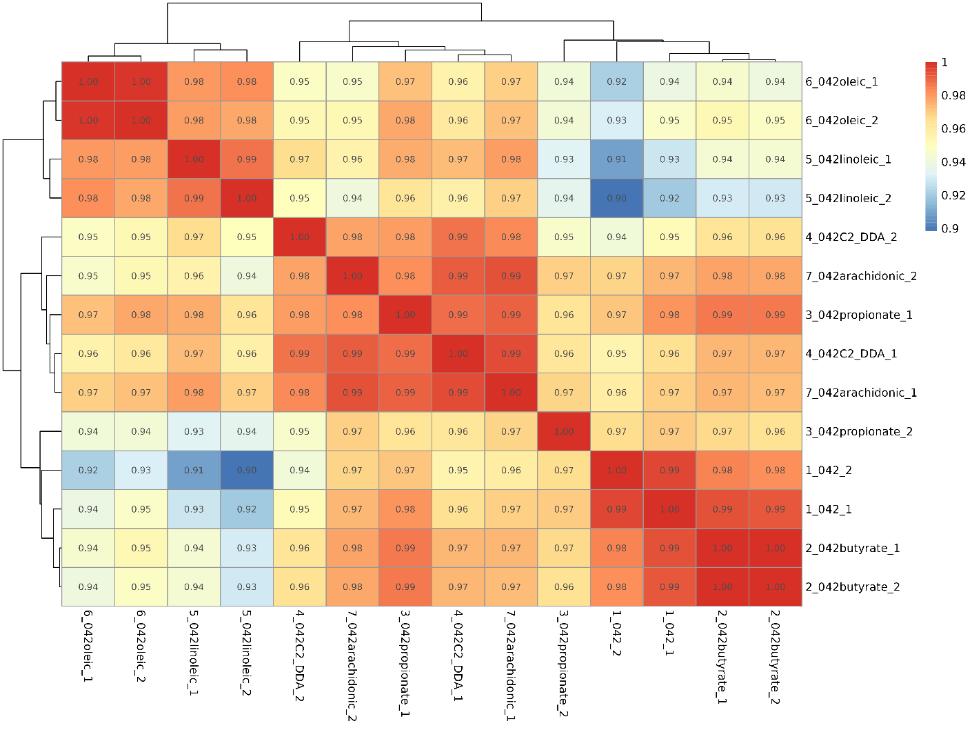
Korrelationsanalyse zwischen Proben
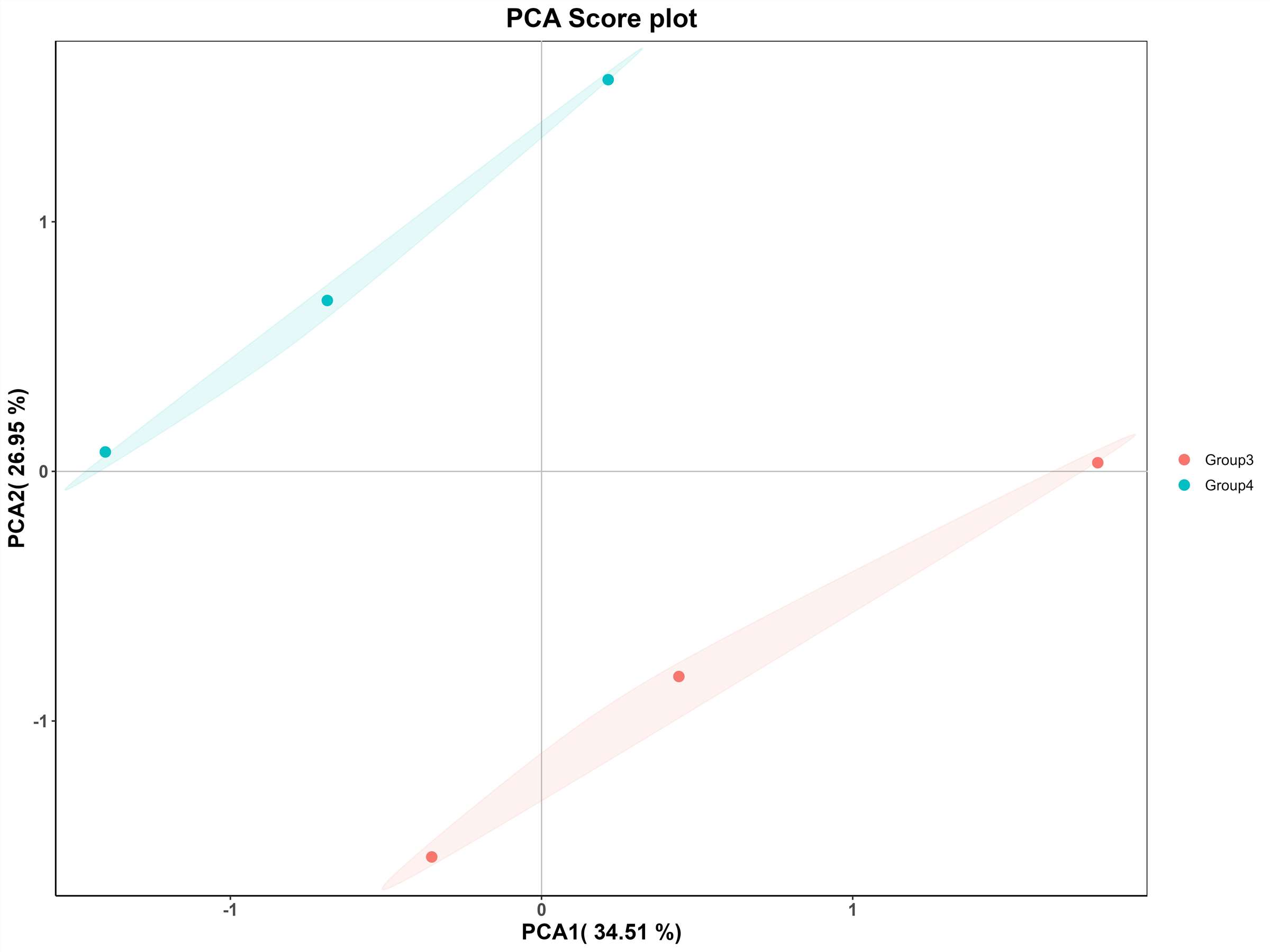
PCA-Score-Diagramm

Venn-Diagramm
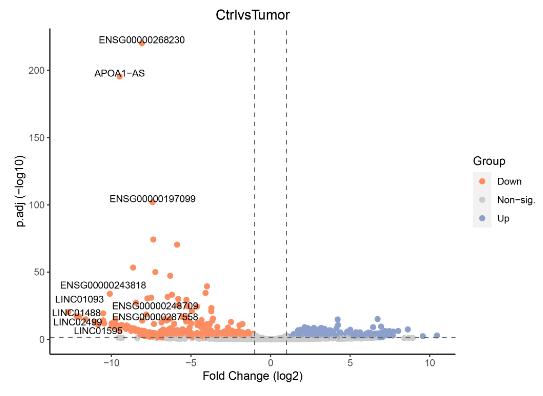
Volcano-Diagramm
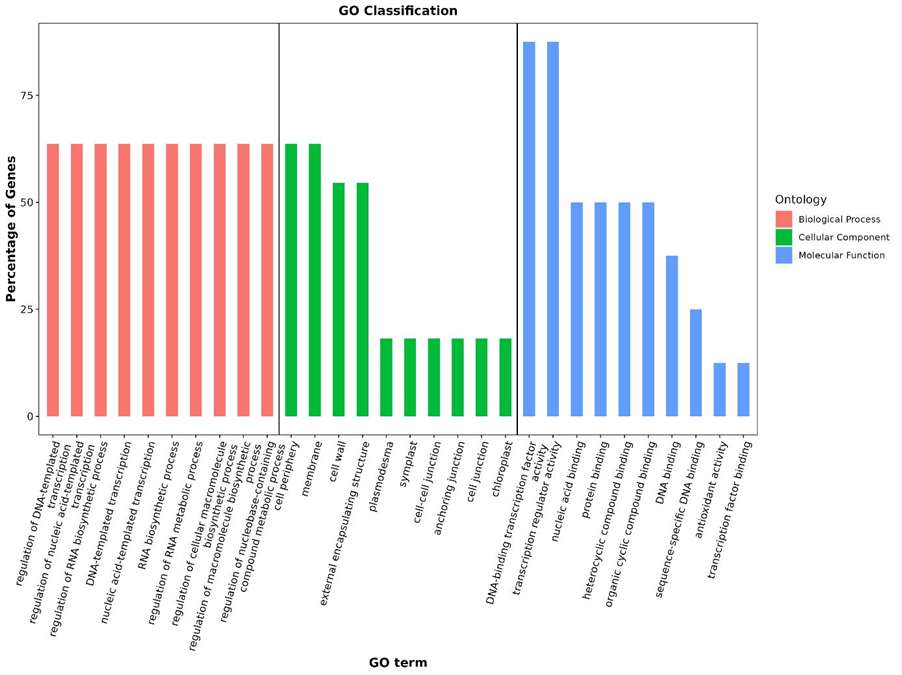
Statistik Ergebnisse der GO-Annotation
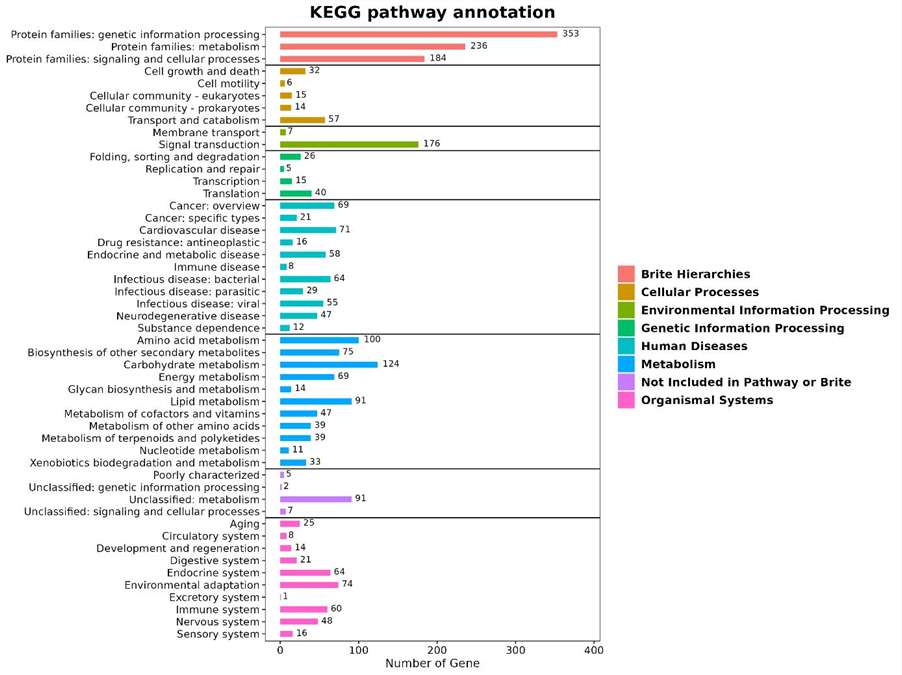
KEGG-Klassifikation
Häufig gestellte Fragen zu Total RNA-Sequenzierung
1. Was wählen: totale RNA-Sequenzierung oder mRNA-Sequenzierung?
Bei der Entscheidung, ob man sich für total RNA-seq oder mRNA-seq für RNA-SequenzierungDie Wahl hängt hauptsächlich von den experimentellen Zielen ab, da es mehrere entscheidende Unterschiede zwischen den beiden Ansätzen gibt. Total RNA-seq, auch bekannt als Whole Transcriptome Sequencing, ist die umfassendste Methode, die typischerweise die Sequenzierung aller Arten von RNA-Molekülen, sowohl kodierend als auch nicht-kodierend, umfasst. Die Entfernung von rRNA während des Total RNA-seq ermöglicht die Erzeugung von hochwertigen Sequenzierungsdaten und erleichtert die Charakterisierung und Identifizierung verschiedener nicht-rRNA-Spezies.
Wenn der Forschungsschwerpunkt auf Eukaryoten liegt und hauptsächlich die kodierenden Regionen betrifft, ist mRNA-seq die bevorzugte Wahl. Die mRNA-seq-Methodik verwendet Selektionsverfahren, um poly(A)-RNA anzureichern. Da mRNA nur einen kleinen Teil der gesamten RNA-Moleküle darstellt, ist mRNA-seq die effektivste und kostengünstigste Methode, wenn die Sequenzierung von mRNA allein für die experimentellen Ziele ausreicht.
2. Welche Art der Sequenzierung ist die Whole Transcriptome Sequencing?
Die gesamte Transkriptom-Sequenzierung verwendet überwiegend Plattformen der zweiten Generation für Kurzlesungen wie Illumina HiSeq X und Illumina NovaSeq 6000. Während die dritte Generation Langzeit-Sequenzierung Es wurden Methoden berichtet, die vorherrschende Wahl für RNA-Seq Experimente bleiben Technologien der zweiten Generation.
3. Was ist das Hauptmerkmal der Whole Transcriptome Sequencing, das sie von anderen Sequenzierungsansätzen unterscheidet?
Das Hauptmerkmal der gesamten Transkriptomsequenzierung liegt in der Verwendung von strangspezifischen Bibliotheksvorbereitungstechniken, insbesondere der strangspezifischen RNA-Bibliotheksvorbereitung. Diese Methodik stellt sicher, dass die resultierenden cDNA-Bibliotheken die Richtungsinformation der ursprünglichen RNA-Stränge bewahren. Dieses Merkmal ist entscheidend für die genaue Bestimmung der transkriptionalen Richtung, wodurch eine präzise Quantifizierung der Genexpression, eine robuste Genannotation, die Entdeckung neuer Gene und eine zuverlässige Identifizierung von Antisense-Transkripten sowie alternativen Spleißereignissen ermöglicht wird.
4. Wie trägt die gesamte Transkriptom-Sequenzierung zum Verständnis der Genregulation und der funktionellen Genomik bei?
Die gesamte Transkriptom-Sequenzierung revolutioniert unser Verständnis von Genregulation und funktioneller Genomik, indem sie eine ganzheitliche Perspektive des Transkriptoms bietet. Diese hochmoderne Technologie taucht in die komplexen Dynamiken der Genexpression ein, untersucht die Feinheiten des alternativen Spleißens und deckt die vielfältigen Signaturen nicht-kodierender RNAs auf, die in verschiedenen biologischen Kontexten vorhanden sind. Die Integration von strangspezifischen RNA-Seq-Daten mit genomischen und epigenomischen Landschaften ausgestattet Forscher mit den Werkzeugen, um die Komplexität regulatorischer Netzwerke zu entschlüsseln und kritische molekulare Akteure in zellulären Prozessen, der Krankheitsentstehung und Entwicklungsabläufen zu enthüllen. Durch die Förderung eines tiefen Verständnisses biologischer Systeme treibt dieser Ansatz Durchbrüche an der Spitze der biomedizinischen Forschung voran und fördert Innovationen sowie bedeutende Entdeckungen.
Gesamt-RNA-Seq Fallstudien
Die Ganztranskriptomsequenzierung zeigt die transkriptionale Landschaft von Neutrophilen, die mit aktiver Tuberkulose assoziiert ist.
Zeitschrift: Frontiers in Immunology
Impact-Faktor: 8,786
Veröffentlicht: 18. August 2022
Zusammenfassung
Neutrophile spielen eine doppelte Rolle bei Tuberkulose: Sie tragen zur angeborenen Immunität gegen M. tuberculosis bei, können jedoch auch Entzündungen und die Pathogenese verschärfen. Transkriptom-Sequenzierung Die Analyse von Neutrophilen liefert Einblicke in ihre komplexen Reaktionen, was das Verständnis der TB-Pathogenese unterstützt und potenzielle Biomarker für die Diagnose identifiziert.
Materialien & Methoden
Probenvorbereitung
Aktive TB-Proben
5 ml peripheres Blut
RNA-Extraktion
Sequenzierung
Bibliotheksbau
RNA-Seq
Illumina NovaSeq6000
DEGs-Analyse
Hauptkomponentenanalyse (PCA)
Genomontologie (GO)
KEGG
Gewichtete Gen-Koexpressionsnetzwerkanalyse (WGCNA)
Ergebnisse
In dieser Studie zeigte die PCA unterschiedliche Genexpressionsprofile zwischen aktiver Tuberkulose (ATB), latenter Tuberkuloseinfektion (LTBI) und gesunden Kontrollgruppen (HC). Die Analyse der differentiellen Genexpression identifizierte signifikante Unterschiede, mit 183 DEGs in ATB vs. HC und 271 DEGs in ATB vs. LTBI. Geschlechtsspezifische Ausdrucksmuster wurden für ATB-herunterregulierte Gene in den Gruppen beobachtet.
 Abbildung 1 Analyse der differentiellen Expression von Neutrophilen in drei Gruppen von Proben.
Abbildung 1 Analyse der differentiellen Expression von Neutrophilen in drei Gruppen von Proben.
Die Autoren führten funktionelle Pfadanreicherung und GSEA-Analysen durch, um die Rollen der differentiell exprimierten Gene (DEGs) zu untersuchen. Die Anreicherungsanalyse ergab, dass Pfade wie die Signalübertragung durch NOD-ähnliche Rezeptoren, die NF-kappa B-Signalübertragung und die Signalübertragung der Immunantwort im Vergleich zu den Gruppen LTBI und HC signifikant in ATB angereichert waren. GSEA hob zusätzliche Pfade wie die Reaktion auf Lipopolysaccharide und die Chemokin-Signalübertragung hervor. Die Analyse des Protein-Interaktionsnetzwerks identifizierte zentrale Subnetzwerke, die die Signalübertragung der Immunantwort und das mRNA-Spleißen betreffen, die insbesondere in den Vergleichen zwischen ATB und LTBI deutlich unterschiedlich waren.
 Abbildung 2 Funktionelle Anreicherungsanalyse von differentiell exprimierten Genen.
Abbildung 2 Funktionelle Anreicherungsanalyse von differentiell exprimierten Genen.
Die KEGG-Analyse zeigte eine signifikante Anreicherung des NF-κB-Signalwegs in der ATB-Gruppe, mit einer Herunterregulierung von TNFSF14 und einer Hochregulierung von NF-kappa B-Inhibitoren (IκBs) wie NFKBIA, NFKBID und NFKBIZ. Darüber hinaus wurden TNFAIP3 und NFKB1A hochreguliert, was möglicherweise die Aktivierung des NF-κB-Signalwegs hemmt. BCL2A1 und CXCL8, die mit dem Überleben von Zellen assoziiert sind, wurden ebenfalls signifikant hochreguliert. Im Gegensatz dazu waren RNA-Spleißungs-bezogene Signalwege in LTBI im Vergleich zu HC herunterreguliert, wie durch GO-, KEGG- und Reactome-Anreicherungsanalysen angezeigt. Die WGCNA-Analyse identifizierte Module, die positiv und negativ mit ATB korreliert waren, und hob unterschiedliche Gene-Co-Expressionsmuster hervor, die mit den TB-Infektionsstatus, insbesondere in Neutrophilen, in Verbindung standen.
 Abbildung 3 Die Hochregulation von NFKB-Inhibitoren in ATB könnte zur Hemmung der Aktivierung des NFKB-Signalwegs führen.
Abbildung 3 Die Hochregulation von NFKB-Inhibitoren in ATB könnte zur Hemmung der Aktivierung des NFKB-Signalwegs führen.
 Abbildung 4 GO- und Reactome-Pfad-Anreicherungsanalyse in LTBI vs. HC im GSEA-Anreicherungsdiagramm.
Abbildung 4 GO- und Reactome-Pfad-Anreicherungsanalyse in LTBI vs. HC im GSEA-Anreicherungsdiagramm.
 Abbildung 5 (A) Die Heatmap der Modul-Eigenschaften-Korrelationsmatrix von WGCNA.
Abbildung 5 (A) Die Heatmap der Modul-Eigenschaften-Korrelationsmatrix von WGCNA.
Neun Zielgene wurden in 76 Proben durch qPCR validiert, wobei eine signifikant höhere Expression von GBP5, SRSF5, CSRNP1, RBM3 und CCNL1 bei ATB festgestellt wurde. Die ROC-Analyse von GBP5, SRSF5, CSRNP1 und RBM3 in 143 Proben bestätigte ihr diagnostisches Potenzial für ATB, wobei GBP5 die stärkste diskriminative Fähigkeit aufwies (AUC>0,9).
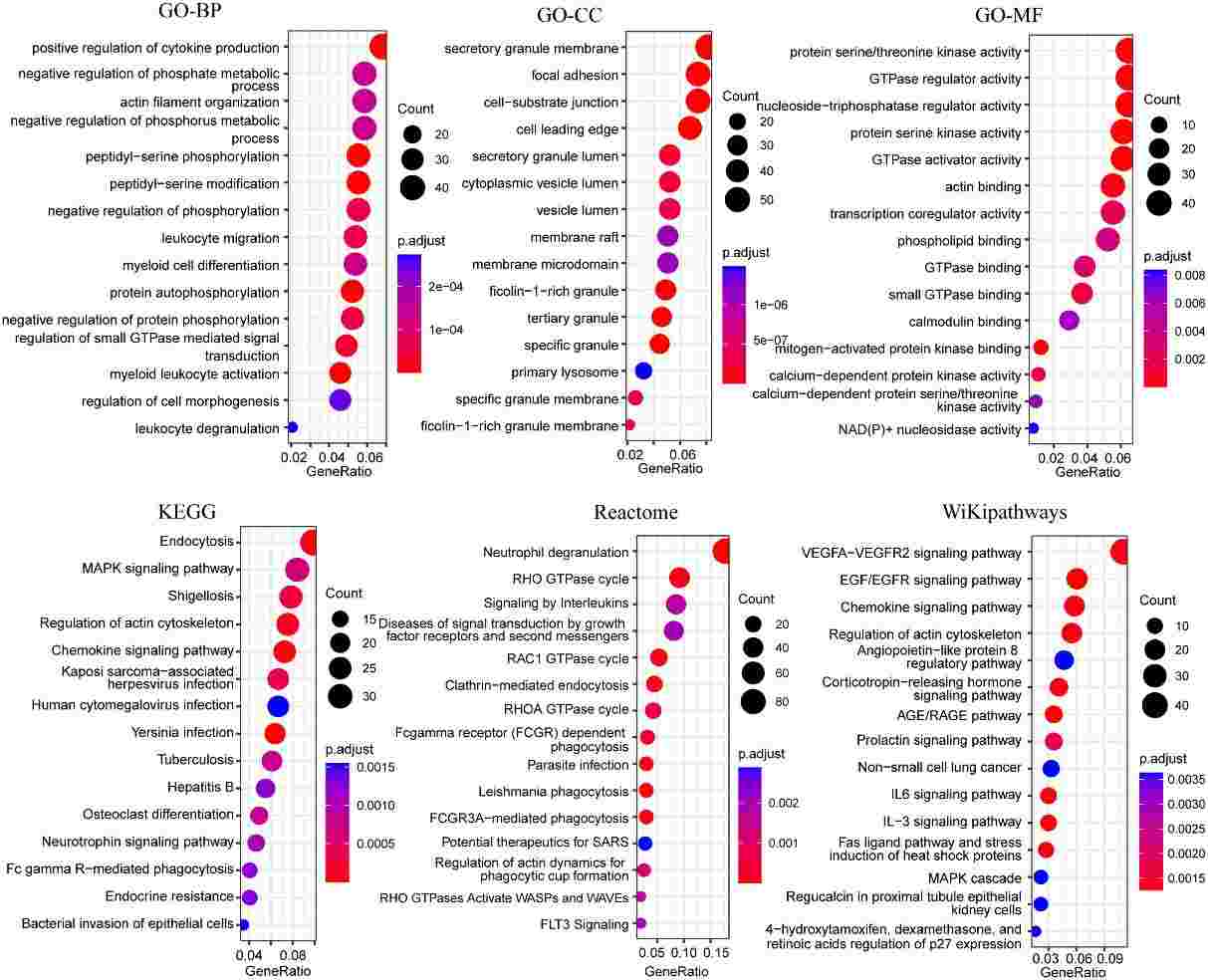 Abbildung 6 Blasendiagramme der funktionellen Anreicherungsanalyse von Genen in blauen Modulen, die nur die 15 am stärksten angereicherten Wege anzeigen.
Abbildung 6 Blasendiagramme der funktionellen Anreicherungsanalyse von Genen in blauen Modulen, die nur die 15 am stärksten angereicherten Wege anzeigen.
Fazit
Diese Studie verwendet Hochdurchsatz-Sequenzierungstechnologie in Verbindung mit umfassenden Analysen, um die Rolle von Neutrophilen als eine der häufigsten Zellen des angeborenen Immunsystems im Prozess der aktiven Tuberkuloseinfektion zu erläutern. Durch die Erkennung von pathogenassoziierten molekularen Mustern über eine Reihe von Mustererkennungsrezeptoren lösen Neutrophile nachgelagerte zelluläre Ereignisse in der Wirtabwehr aus, insbesondere in Bezug auf immunantwortinduzierte Signalwege. Mycobacterium tuberculosis kann jedoch die antibakterielle Funktion von Neutrophilen beeinträchtigen, indem es den NF-kB-Signalweg hemmt, wodurch Entzündungen und Zellapoptose abgeschwächt und ihr Überleben innerhalb der Neutrophilen gefördert wird. Durch RT-qPCR-Analysen wurden drei neuartige transkriptionale Marker-Gene, RBM 3, CSRNP 1 und SRSF 5, identifiziert, die das Potenzial haben, zwischen aktiver Tuberkulose, latenter Tuberkuloseinfektion und gesunden Kontrollen zu unterscheiden.
Referenz
- Geng X, Wu X, Yang Q, et al. Die Ganztranskriptom-Sequenzierung zeigt die transkriptionale Landschaft von Neutrophilen, die mit aktiver Tuberkulose assoziiert ist. Grenzen der Immunologie, 2022, 13: 954221.
Verwandte Veröffentlichungen
Hier sind einige Veröffentlichungen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Chaperon-vermittelte Autophagie steuert proteomische und transkriptomische Wege, um die Aktivität von Gliom-Stammzellen aufrechtzuerhalten.
Journal: Krebsforschung
Jahr: 2022
Zirkuläre DNA-Tumorviren erzeugen zirkuläre RNAs.
Zeitschrift: Mitteilungen der Nationalen Akademie der Wissenschaften
Jahr: 2018
Wiederholte Immunisierung mit ATRA-haltigem liposomalem Adjuvans transdifferenziert Th17-Zellen zu einem Tr1-ähnlichen Phänotyp.
Zeitschrift: Zeitschrift für Autoimmunität
Jahr: 2024
Die Rolle der Histonvariante H2A.Z.1 bei Gedächtnis, Transkription und alternativer Spleißung wird durch Lysinmodifikationen vermittelt.
Zeitschrift: Neuropsychopharmakologie
Jahr: 2024
FAK-Verlust reduziert die ERK-Phosphorylierung, die durch BRAFV600E induziert wird, um die intestinale Stammzelligkeit und die Bildung von Blinddarmtumoren zu fördern.
Journal: Elife
Jahr: 2023
Identifizierung von zirkulären RNAs, die die Proliferation von Kardiomyozyten in neonatalen Schweineherzen regulieren
Journal: JCI Insight
Jahr: 2024
Mehr ansehen Artikel, die von unseren Kunden veröffentlicht wurden.


