
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
LncRNA-Sequenzierung
CD Genomics bietet einen hochdurchsatzfähigen und kosteneffizienten lncRNA-Sequenzierungsdienst an, indem die neuesten Illumina-Sequenzierungsinstrumente und fortschrittliche bioinformatische Analysen kombiniert werden.
Die Einführung der LncRNA-Sequenzierung
Lange nicht-kodierende RNAs (lncRNAs) werden als eine große und vielfältige Klasse transkribierter RNAs definiert, die größer als 200 nt sind und keine Proteine kodieren. LncRNAs sind in Organismen weit verbreitet, und lncRNA-Transkripte machen den größten Teil des nicht-kodierenden Transkriptoms aus. LncRNAs können in verschiedene Subtypen (einschließlich Antisense-, intergenische, überlappende, intronische, bidirektionale und prozessierte) eingeteilt werden, basierend auf der Position und Richtung der Transkription in Bezug auf andere Gene. LncRNAs ähneln mRNAs, da sie typischerweise aus aktivem Chromatin transkribiert, polyadenyliert und gekappt werden. Sie leiten jedoch nicht die Proteinsynthese.
LncRNAs sind funktional wichtig für Organismen und nicht nur das Produkt transkriptioneller Geräusche. Eine Vielzahl molekularer Funktionen von lncRNAs wurde bei Säugetieren und Pflanzen entdeckt, einschließlich der Neupositionierung von Nukleosomen, Chromatinumbau, transkriptioneller Kontrolle und posttranskriptioneller Verarbeitung. LncRNAs werden zunehmend mit dem Auftreten von Krankheiten, genomischer Prägung und Entwicklungsregulation in Verbindung gebracht. Die Genexpressionsprofilierung und in situ Hybridisierungsstudien haben gezeigt, dass die Expression von lncRNA entwicklungsbedingt reguliert ist, gewebespezifisch und zellspezifisch sein kann und räumlich, zeitlich oder als Reaktion auf Stimuli variieren kann.
Die Anwendung von Next-Generation-Sequenzierung Die Technologie hat die Entdeckung und Funktionsanalyse von lncRNAs erheblich erleichtert. Informationen über lncRNA-Sequenzen können mit einer Einzelbasenauflösung durch Bibliotheksvorbereitung, Hochdurchsatz-Sequenzierung und leistungsstarke Bioinformatik Analyse. Wir erstellen die Sequenzierungsbibliothek durch die Entfernung von rRNA und behalten sowohl lncRNAs als auch mRNAs. Die Analyse der lncRNA-mRNA-Interaktionen trägt zur Aufklärung der lncRNA-Regulationsnetzwerke bei.
Vorteile der LncRNA-Sequenzierung
- Identifiziert bekannte und neue Merkmale
- Ermöglicht die Profilerstellung von lncRNAs über ein breites dynamisches Spektrum.
- Untersucht neuartige Biomarker und lncRNA-Regulationsnetzwerke
Anwendungen der LncRNA-Sequenzierung
Die LncRNA-Sequenzierung kann für, aber nicht beschränkt auf, die folgenden Forschungsbereiche verwendet werden:
- Entdeckung und Vorhersage neuartiger lncRNAs;
- Krankheitsuntersuchung;
- Regulatorische Netzwerk Analyse.
LncRNA-Sequenzierungs-Workflow
Der allgemeine Arbeitsablauf für lncRNA-Sequenzierung ist unten skizziert. Um eine lncRNA-Sequenzierungsbibliothek zu erstellen, besteht der erste Schritt der lncRNA-Sequenzierung darin, rRNA zu entfernen, gefolgt von RNA-Fragmente, cDNA-Synthese, Adaptor-Ligation, Größen-Selektion und PCR-Anreicherung. Unser hochqualifiziertes Expertenteam führt das Qualitätsmanagement durch und überwacht jeden Schritt, um zuverlässige und unvoreingenommene Ergebnisse zu gewährleisten.

Dienstspezifikation
Musteranforderungen
|
|
 Klicken |
Sequenzierungsstrategien
|
 |
Datenanalyse Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an:
|
Analyse-Pipeline

Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details zur LncRNA-Sequenzierung für Ihre Schreibanpassung.
Unser Bioinformatik-Team auf Doktoratsniveau bietet umfassende Analysen für sowohl lncRNAs als auch mRNAs und ermöglicht den Zugriff auf lncRNA- und mRNA-Informationen in einem einzigen Sequenzierungslauf. Wir können Ihnen bei der experimentellen Planung zu Beginn Ihres Projekts helfen und bieten Beratung in jeder Phase des Projektprozesses an.
Teilweise Ergebnisse sind unten aufgeführt:

Sequenzierungsqualitätsverteilung
A/T/G/C-Verteilung
IGV-Browser-Oberfläche
Korrelationsanalyse zwischen Proben
PCA-Score-Diagramm

Venn-Diagramm
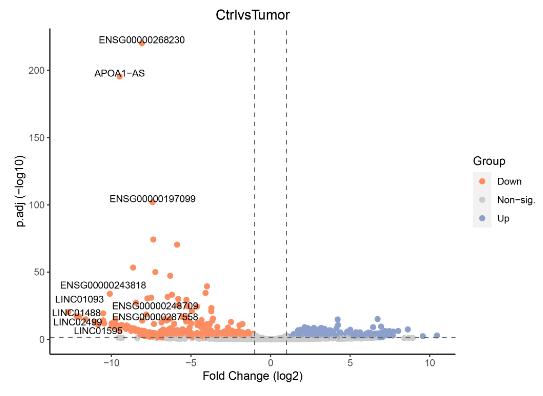
Volcano-Diagramm
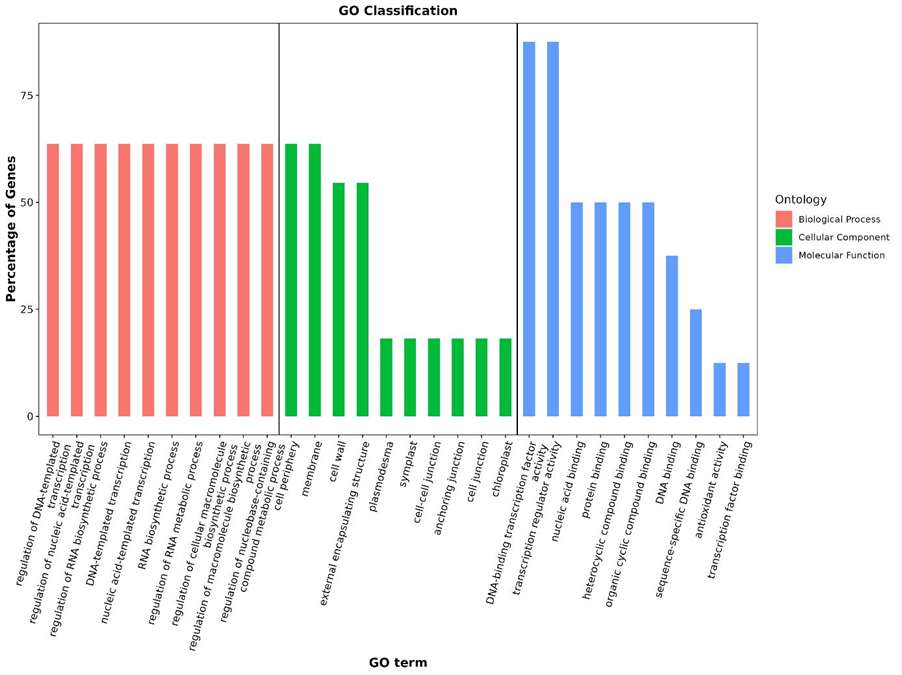
Statistiken der Ergebnisse der GO-Annotation
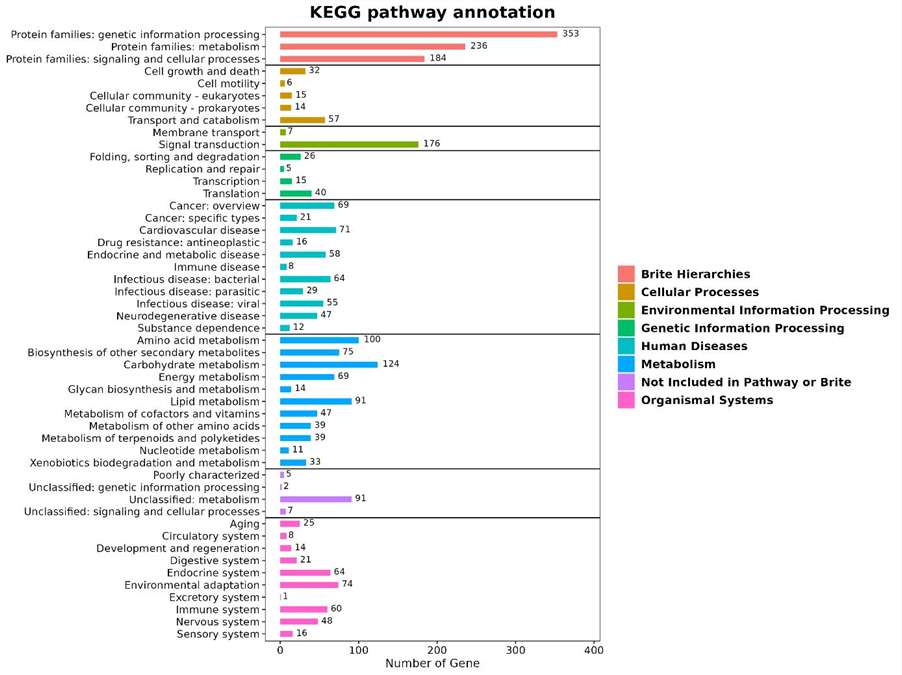
KEGG-Klassifikation
1. Welche Arten sind für lncRNA-Sequenzierungsstudien geeignet?
Für lncRNA-Sequenzierungsstudien müssen die geeigneten Arten die folgenden Anforderungen erfüllen: (i) Eukaryoten; (ii) mindestens ein Referenzgenom auf Scaffold-Ebene verfügbar; (iii) relativ vollständige Genomanalysen.
2. Warum rRNA entfernen, wenn man lncRNA-Sequenzierungsbibliotheken erstellt?
Ribosomale RNA (rRNA) ist der am häufigsten vorkommende Bestandteil der gesamten RNA und macht 80 % bis 90 % der Moleküle in einer gesamten RNA-Probe von Tieren oder Menschen aus. Um eine effiziente Transkriptdetektion zu gewährleisten, müssen die hochabundanten rRNAs vor der Sequenzierung entfernt werden.
3. Wie man lncRNA-Ziele vorhersagt?
Beide Analysen der Ko-Lokalisierung und Ko-Expression von protein-kodierenden RNAs und lncRNAs haben sich als effektiv in den Untersuchungen der potenziellen Funktion von lncRNAs in biologischen Prozessen und der Vorhersage von lncRNA-Zielen erwiesen. Während die Ko-Lokalisierungsanalyse die benachbarten kodierenden Gene berücksichtigt, die möglicherweise lncRNA-Ziele sind, betrachtet die Ko-Expressionsanalyse die ko-exprimierten Gene als wahrscheinliche lncRNA-Ziele, unabhängig von ihrem Standort.
4. Was sind die Unterschiede zwischen lncRNA und mRNA?
LncRNAs ähneln mRNAs da sie typischerweise aus aktiver Chromatin transkribiert, polyadenyliert und gekappt werden. Sie leiten jedoch nicht die Proteinsynthese ein. Einige Unterschiede zwischen lncRNA und mRNA sind in der folgenden Tabelle zusammengefasst.
| mRNA | lncRNA |
| Protein-kodierendes Transkript | Nicht-protein-kodierende, regulatorische Transkripte |
| Gut zwischen den Arten erhalten | Schlecht zwischen den Arten erhalten. |
| In sowohl im Zellkern als auch im Zytoplasma vorhanden | Viele sind überwiegend nuklear, andere nuklear und/oder zytoplasmatisch. |
| Insgesamt 20-24.000 mRNAs | Derzeit gibt es etwa 30.000 lncRNA-Transkripte, die in ihrer Anzahl auf das 3- bis 100-fache von mRNA geschätzt werden. |
| Ausdrucksniveau: niedrig bis hoch | Ausdrucksniveau: sehr niedrig bis moderat |
Identifizierung von islet-angereicherten langen nicht-kodierenden RNAs, die zum β-Zell-Versagen bei Typ-2-Diabetes beitragen
Journal: Molekulare Metabolismus
Impact-Faktor: 6,799
Veröffentlicht: 23. August 2017
Zusammenfassung
Die Autoren identifizierten ungefähr 1500 neuartige lncRNAs, von denen einige in fettleibigen Mäusen unterschiedlich exprimiert wurden. Zwei lncRNAs (βlinc2 und βlinc3) sind in β-Zellen stark angereichert, korreliert mit der Zunahme des Körpergewichts und den Glykemiewerten in fettleibigen Mäusen und verändert in diabetischen db/db-Mäusen. Darüber hinaus wurde die Expression des menschlichen Orthologen von βlinc3 in den Inseln von Patienten mit Typ-2-Diabetes verändert, was mit dem BMI der Spender assoziiert war. Die Modulation des Levels der beiden lncRNAs durch Überexpression oder Herunterregulierung in MIN6- und Mausinsulinzellen erhöhte die β-Zellen, hatte jedoch keinen Einfluss auf die Glukoseregulation.
Materialien & Methoden
- Fünf Wochen alte männliche C57BL/6-Mäuse
- Mausinseln
- RNA-Extraktion
- Bibliothekskonstruktion
- RNA-Sequenzierung
- Illumina HiSeq2000
- Differenzielle Expressionsanalyse
- Differentialanalyse
- Genomontologie-Analyse
- Messung der lncRNA-Expression
Ergebnisse
1. RNA-Sequenzierungsanalyse
Die RNA-Sequenzierung ergab 1558 neuartige lncRNAs, von denen einige in fettleibigen Mäusen unterschiedlich exprimiert wurden. Die funktionelle Annotation zeigte eine Anreicherung für biologische Signalwege, die mit Proteinlokalisierung und -transport, Redoxprozessen sowie intrazellulärem Transport in Verbindung stehen.
 Abbildung 1. Übersicht der RNA-Sequenzierungsergebnisse. (A) Hierarchische Clusteranalyse. Rot steht für keine Distanz und Gelb für eine größere Distanz. ND steht für normale Ernährung, HDR steht für Hochfettkost-Responder. (B) Zusammenfassung der differentially exprimierten Gene. (C) Kodierungspotenzial für neuartige Transkripte; (D) Größenverteilung von protein-codierenden Genen, bekannten und neuartigen lncRNAs. (E & F) Locus-Architektur und Isoformen von βlinc2 oder βlinc3.
Abbildung 1. Übersicht der RNA-Sequenzierungsergebnisse. (A) Hierarchische Clusteranalyse. Rot steht für keine Distanz und Gelb für eine größere Distanz. ND steht für normale Ernährung, HDR steht für Hochfettkost-Responder. (B) Zusammenfassung der differentially exprimierten Gene. (C) Kodierungspotenzial für neuartige Transkripte; (D) Größenverteilung von protein-codierenden Genen, bekannten und neuartigen lncRNAs. (E & F) Locus-Architektur und Isoformen von βlinc2 oder βlinc3.
2. Die Expressionsniveaus von βlinc2 und βlinc3
 Abbildung 2. Die Expressionsniveaus von βlinc2 und βlinc3 sind in Inseln von Mäusen, die eine fettreiche Diät erhalten haben, und in db/db-Mäusen verändert.
Abbildung 2. Die Expressionsniveaus von βlinc2 und βlinc3 sind in Inseln von Mäusen, die eine fettreiche Diät erhalten haben, und in db/db-Mäusen verändert.
 Abbildung 3. Korrelationen der Expression von βlinc2 und βlinc3 mit dem Körpergewicht und der Glykämie von C57BL/6-Mäusen.
Abbildung 3. Korrelationen der Expression von βlinc2 und βlinc3 mit dem Körpergewicht und der Glykämie von C57BL/6-Mäusen.
 Abbildung 4. In vitro Auswirkungen von chronisch erhöhtem Glukose- und Palmitatgehalt auf das Niveau der beiden lncRNAs, die in Inseln von Mäusen, die eine fettreiche Diät erhalten haben, unterschiedlich exprimiert werden.
Abbildung 4. In vitro Auswirkungen von chronisch erhöhtem Glukose- und Palmitatgehalt auf das Niveau der beiden lncRNAs, die in Inseln von Mäusen, die eine fettreiche Diät erhalten haben, unterschiedlich exprimiert werden.
 Abbildung 5. Die βlinc3-Expression ist in Inseln von Spendern mit Typ-2-Diabetes verringert.
Abbildung 5. Die βlinc3-Expression ist in Inseln von Spendern mit Typ-2-Diabetes verringert.
 Abbildung 6. Überexpression und Herunterregulierung von βlinc2 und βlinc3 fördern die Apoptose in MIN6 β-Zellen. (A) MIN6-Zellen wurden mit einem Kontrollvektor oder Plasmiden transfiziert, um die Überexpression von βlinc2 oder βlinc3 zu induzieren. (B) Die Zellen wurden mit einem Kontroll-Gapmer oder einem Gapmer transfiziert, um βlinc3 herunterzuregulieren. (C & D) sind die wiederholten Experimente in dispergierten Mausinseln. (E) MIN6-Zellen wurden mit einem Plasmid transfiziert, das p65 mit GFP-Tag exprimiert.
Abbildung 6. Überexpression und Herunterregulierung von βlinc2 und βlinc3 fördern die Apoptose in MIN6 β-Zellen. (A) MIN6-Zellen wurden mit einem Kontrollvektor oder Plasmiden transfiziert, um die Überexpression von βlinc2 oder βlinc3 zu induzieren. (B) Die Zellen wurden mit einem Kontroll-Gapmer oder einem Gapmer transfiziert, um βlinc3 herunterzuregulieren. (C & D) sind die wiederholten Experimente in dispergierten Mausinseln. (E) MIN6-Zellen wurden mit einem Plasmid transfiziert, das p65 mit GFP-Tag exprimiert.
Fazit
Dieses Papier identifizierte eine große Anzahl neuartiger lncRNAs. Mindestens zwei von ihnen können das Überleben von β-Zellen beeinflussen und könnten zur glucolipotoxischen Mediierung von β-Zellen sowie zur Manifestation und Progression von T2D beitragen. LncRNA könnte daher die idealen Ziele für die Prävention und Behandlung von Diabetes bieten.
Referenz:
- Motterle A, Gattesco S, Peyot M L, u. a.Identifizierung von islet-reichen langen nicht-kodierenden RNAs, die zum β-Zell-Versagen bei Typ-2-Diabetes beitragen. Molekularer Stoffwechsel, 2017, 6(11): 1407-1418.
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Die Spaltung von Phagen-DNA durch Restriktionsendonukleasen ermöglicht die Wiederbelebung aus der durch Cas13 induzierten bakteriellen Dormanz.
Zeitschrift: Nature Mikrobiologie
Jahr: 2023
IL-4 fördert die Erschöpfung von CD8.+ CART-Zellen
Zeitschrift: Nature Communications
Jahr: 2024
Fettreiche Diäten während der Schwangerschaft führen zu Veränderungen der DNA-Methylierung und Proteinexpression im Pankreasgewebe des Nachwuchses: Ein Multi-Omics-Ansatz
Internationale Zeitschrift für Molekulare Wissenschaften
Jahr: 2024
KMT2A assoziiert mit dem PHF5A-PHF14-HMG20A-RAI1 Subkomplex in Stammzellen des Bauchspeicheldrüsenkrebses und reguliert epigenetisch deren Eigenschaften.
Zeitschrift: Nature Communications
Jahr: 2023
Krebsassoziierte DNA-Hypermethylierung von Polycomb-Zielen erfordert die doppelte Erkennung von Histon H2AK119-Ubiquitinierung und der sauren Tasche des Nukleosoms durch DNMT3A.
Journal: Wissenschaftliche Fortschritte
Jahr: 2024
Genomisches Imprinting-ähnliches monoalleles väterliches Ausdrucksmuster bestimmt das Geschlecht von Kanalkatzenfischen.
Journal: Wissenschaftliche Fortschritte
Jahr: 2022
Mehr ansehen Artikel, die von unseren Kunden veröffentlicht wurden.


