
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Genomweite Assoziationsstudie (GWAS)
Die Einführung von GWAS
Genomweite Assoziationsstudien (GWAS) haben die genetische Forschung revolutioniert, indem sie eine robuste Methodik zur Identifizierung genetischer Variationen bereitstellen, die mit spezifischen Merkmalen und Krankheiten korreliert sind. Durch umfassendes Scannen der Genome über große Populationen hinweg versucht GWAS, die Beziehung zwischen genetischen Varianten und phänotypischen Merkmalen zu entschlüsseln. Dieser methodische Ansatz offenbart tiefgreifende Einblicke in die genetischen Grundlagen verschiedener Erkrankungen, verbessert unser Verständnis der menschlichen Genetik erheblich und ebnet den Weg für die Entwicklung gezielter therapeutischer Interventionen.
Was ist der Zweck von GWAS?
GWAS sind eine leistungsstarke Methodik, die darauf abzielt, genetische Varianten zu identifizieren, die mit komplexen Krankheiten und Merkmalen assoziiert sind. Im Gegensatz zu traditionellen familienbasierten Verknüpfungsstudien nutzt GWAS Hochdurchsatz- Genotypisierung um umfassende Scans über das gesamte Genom durchzuführen. Dieser Ansatz ermöglicht es Forschern, Einzelne Nukleotid-Polymorphismen (SNPs) und andere genetische Marker zu erkennen, die mit Erkrankungen wie Diabetes, Herz-Kreislauf-Erkrankungen und verschiedenen Krebsarten korreliert sind.
Hauptziele von GWAS
- Identifizierung von krankheitsassoziierten VariantenDas Hauptziel von GWAS besteht darin, spezifische genetische Varianten aufzudecken, die das Risiko für Krankheiten erhöhen. Zum Beispiel verbessert die Identifizierung von SNPs, die mit der Anfälligkeit für Brustkrebs assoziiert sind, unser Verständnis von genetischen Risikofaktoren und ebnet den Weg für gezielte therapeutische Interventionen.
- Verständnis der genetischen ArchitekturGWAS bietet Einblicke, wie multiple genetische Varianten zusammenwirken, um komplexe Merkmale zu beeinflussen. Durch die Aufklärung dieser Interaktionen können Forscher die zugrunde liegenden biologischen Mechanismen von Krankheiten und Merkmalen besser verstehen, was zu robusteren Modellen der genetischen Architektur führt.
- Unterstützung der personalisierten MedizinDurch die Identifizierung genetischer Risikofaktoren trägt GWAS erheblich zur Weiterentwicklung der personalisierten Medizin bei. Dieser präzise Ansatz ermöglicht die Anpassung von präventiven und therapeutischen Strategien, die auf individuelle genetische Profile zugeschnitten sind, und verbessert somit die Gesundheitsversorgung.
Vorteile von GWAS
- Umfassende DeckungGWAS scannen das gesamte Genom und erhöhen die Chancen, neue genetische Marker zu entdecken, die mit Krankheiten verbunden sind.
- Hohe Durchsatzrate und SkalierbarkeitFortgeschrittene Genotypisierungstechnologien ermöglichen die Analyse vieler Varianten in großen Populationen und verbessern die Nachweisempfindlichkeit.
- Unvoreingenommene EntdeckungGWAS verwenden einen hypothesenfreien Ansatz, der Verzerrungen verringert und unerwartete genetische Verbindungen zu Krankheiten aufdeckt.
- Reproduzierbarkeit und ValidierungErgebnisse aus GWAS können in unabhängigen Studien validiert werden, um die Zuverlässigkeit und Genauigkeit der Ergebnisse sicherzustellen.
Anwendungen von GWAS
- KrankheitsforschungDie Identifizierung genetischer Varianten, die mit Krankheiten wie Herzkrankheiten und Krebs in Verbindung stehen, einschließlich SNPs, die mit Alzheimer-Krankheit assoziiert sind, hilft, zukünftige therapeutische Entwicklungen zu leiten.
- PharmakogenomikDie Entdeckung genetischer Varianten, die die individuellen Arzneimittelreaktionen beeinflussen, ermöglicht die Entwicklung personalisierter Arzneimitteltherapien, die die Wirksamkeit erhöhen und Nebenwirkungen verringern.
- Agrarische GenetikDas Auffinden genetischer Varianten, die mit vorteilhaften Eigenschaften bei Pflanzen und Tieren, wie Ertrag und Trockenresistenz, in Verbindung stehen, verbessert die landwirtschaftliche Produktivität und Resilienz.
- PopulationsgenetikDas Verständnis genetischer Vielfalt und evolutionärer Geschichte innerhalb von Populationen zeigt, wie Variationen zwischen ethnischen Gruppen die Krankheitsanfälligkeit beeinflussen.
GWAS-Workflow
GWAS umfasst die Auswahl einer Studienpopulation, das Genotypisieren von Individuen zur Identifizierung genetischer Varianten und die Verwendung statistischer Modelle, um Zusammenhänge zwischen diesen Varianten und spezifischen Merkmalen oder Krankheiten zu finden. Die Ergebnisse werden durch Replikation in unabhängigen Kohorten validiert, um ihre Zuverlässigkeit zu bestätigen.

Dienstspezifikationen
Musteranforderungen
|
|
 Klicken |
Sequenzierungsstrategie
|
 |
Bioinformatikanalyse Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an:
|
Analyse-Pipeline
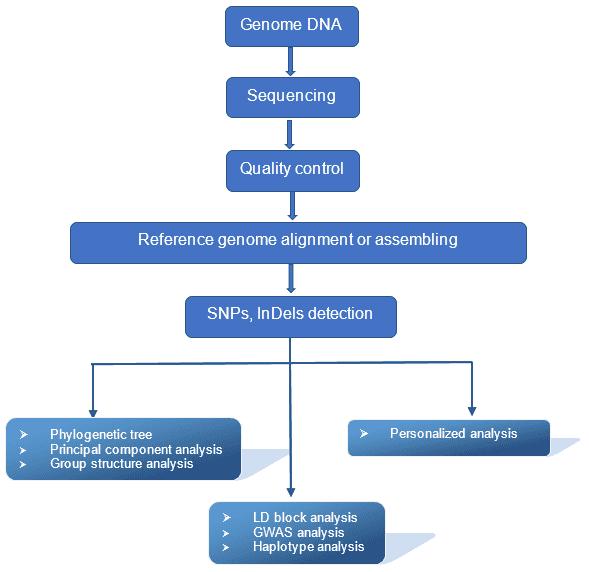
Liefergegenstände
- Rohdaten (FASTQ)
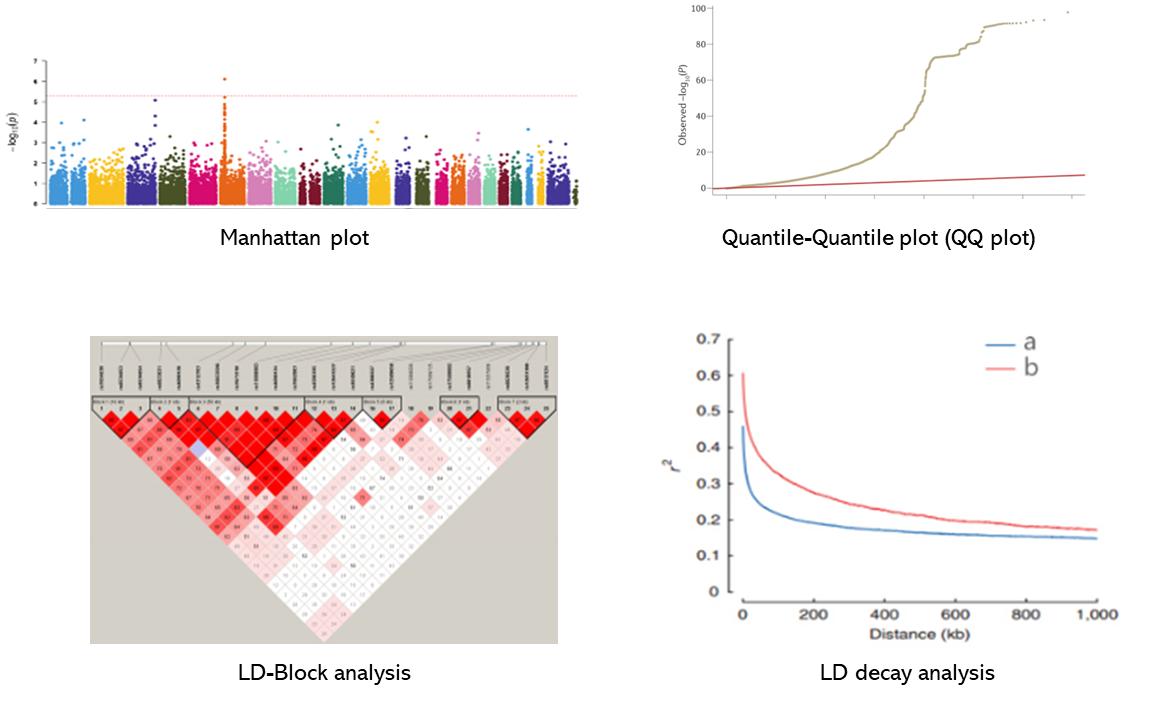
- Bedeutende SNP-Informationen
- QQ-Plot und Manhattan-Plot
- Datenanalysebericht
Demonstrationsergebnisse
Teilweise Ergebnisse werden unten angezeigt:
Häufig gestellte Fragen zu genomweiten Assoziationsstudien
1. Was sind die Prinzipien für die Stichprobenauswahl in genomweiten Assoziationsstudien (GWAS)?
- Sicherstellen einer ausreichenden Repräsentativität der Proben: Die ausgewählten Proben müssen die Zielpopulation ausreichend repräsentieren, um sicherzustellen, dass die Ergebnisse allgemein anwendbar sind.
- Vermeiden Sie Proben mit signifikanter Subpopulationen-Stratifizierung: Proben sollten keine ausgeprägte Differenzierung zwischen Subpopulationen aufweisen (z. B. reproduktive Isolation), da eine solche Stratifizierung erhebliches genetisches Hintergrundrauschen einführen kann, das die Analyse verfälscht.
- Fokus auf Phänotypen mit hoher Erblichkeit: Es ist ratsam, mehrere Schlüsselphänotypmerkmale mit hoher Erblichkeit als primäre Zielsetzungen für die Studie zu priorisieren, um die Wahrscheinlichkeit zu erhöhen, signifikante Assoziationen zu erkennen.
- Nutzen Sie binäre Merkmale für qualitative Eigenschaften: Bei qualitativen Eigenschaften sollten Sie versuchen, binäre Phänotypen (0/1) zu verwenden und sicherzustellen, dass die Stichprobengrößen für die beiden phänotypischen Kategorien ungefähr gleich sind, um robuste statistische Vergleiche zu ermöglichen.
- Quantitative Merkmale genau quantifizieren: Quantitative Merkmale sollten präzise gemessen und aufgezeichnet werden, wie zum Beispiel die Krankheitsresistenz, die durch Inzidenzrate, Sterberate, Überlebensrate, Läsionsanzahl oder Läsionsfläche quantifiziert wird, anstatt breite kategoriale Skalen zu verwenden. Die phänotypischen Daten sollten idealerweise einer nahezu normalen Verteilung folgen.
- Nutzen Sie langfristige Mehrstandortversuche für kultivierte Pflanzen: Im Falle von kultivierten Pflanzen sollten mehrjährige, mehrstandortliche und wiederholte Versuche durchgeführt werden. Die Ergebnisse dieser Versuche können separat analysiert oder gemittelt werden, um die Zuverlässigkeit der Assoziationsanalysen zu stärken.
- Anpassung der Stichprobengröße basierend auf phänotypischer Variabilität und Kontrolle: Wenn die phänotypische Variation erheblich ist und von Hauptloci kontrolliert wird, kann eine kleinere Stichprobengröße (empfohlene Mindestanzahl von 200 Individuen) ausreichen. Bei Merkmalen mit geringen phänotypischen Unterschieden und polygenetischer Kontrolle ist jedoch eine größere Stichprobengröße (empfohlene Mindestanzahl von 500 Individuen) erforderlich, um signifikante Assoziationen zu erkennen.
2. Was sind die Themen von GWAS in natürlichen Populationen?
Nicht-streng genetische Populationen:
- Genressourcen
- Halbgeschwister und gemischte Populationen
- MAGIE/NAM Bevölkerungen
- Mehrere F2/RIL- oder Vollgeschwisterpopulationen
- Hoch heterozygote Arten: F1-Populationen
3. Können verschiedene Merkmale in einer einzelnen Person überlappen?
Ja, unterschiedliche Merkmale können bei derselben Person überlappen. Zum Beispiel können bei der Kategorisierung einer Population basierend auf Größe und Farbmerkmalen Individuen in beiden Gruppen vorhanden sein. Diese Überlappung beeinträchtigt nicht die Gültigkeit der Analyseergebnisse.
4. Ist GWAS ohne ein Referenzgenom möglich?
In Abwesenheit eines Referenzgenoms ermöglichen vereinfachte Genomsequenzierungstechnologien wie RAD-seq oder GBS kann eingesetzt werden, um SNPs durch Clustering zu erkennen. Obwohl diese SNPs für GWAS verwendet werden können, schränkt das Fehlen von Genomannotationen die weitere Genannotierung der identifizierten Assoziationsloci ein.
5. Wie werden GWAS-Ergebnisse validiert?
GWAS-Ergebnisse werden durch Replikationsstudien in unabhängigen Kohorten validiert. Darüber hinaus helfen funktionale Studien und Pfadanalysen, die biologische Relevanz der identifizierten genetischen Varianten zu bestätigen.
Genomweite Assoziationsstudie Fallstudien
Genomweite Assoziations- und Multi-Trend-Analysen charakterisieren die gemeinsame genetische Architektur von Herzinsuffizienz.
Zeitschrift: Nature Communications
Impactfaktor: 16,6
Veröffentlicht: 14. November 2022
Hintergrund
Herzinsuffizienz (HF) betrifft weltweit über 38 Millionen Menschen und ist eine Hauptursache für kardiovaskuläre Probleme. Trotz ihrer hohen Prävalenz wurden nur 11 genetische Loci identifiziert, die mit HF assoziiert sind. Diese Studie verbessert die GWAS-Power, indem sie Daten aus verschiedenen Abstammungen kombiniert und kardiologische Bildgebungsmerkmale integriert. Sie entdeckt neue Risikovarianten für HF, identifiziert relevante Gewebe und untersucht genetische Assoziationen mit zirkulierenden Proteinen und Bildgebungsphänotypen.
Materialien & Methoden
Probenvorbereitung
- Mensch
- Herzinsuffizienz
- Gewebe- und Zelltypanreicherung
Methode
- Meta-Analyse von genomweiten Assoziationsstudien
- Multivariate GWAS
- Transkriptomweite Assoziationsstudie
- Cross-Trait Linkage-Disequilibrium-Score-Regressions (LDSC)
- Multi-Trait-Ko-Lokalisierung
- Kardiake Genexpressionsprofilierung
- Analyse biologischer Wege und zellulärer Komponenten
Ergebnisse
Eine multianalytische Meta-Analyse zu Herzinsuffizienz identifizierte 47 Risikoloci anhand von Daten von über 115.000 HF-Fällen und 1,5 Millionen Kontrollen. Unter diesen erreichten 939 Varianten genomweite Signifikanz, wobei 34 Loci außerhalb zuvor berichteter Regionen gefunden wurden. Die stärkste Assoziation war am PITX2-Locus. Die Replikation in zusätzlichen Kohorten bestätigte 41 von 44 Loci mit übereinstimmenden Effekten. Pleiotropie-Analysen zeigten, dass viele HF-Loci auch mit anderen kardiometabolischen Merkmalen assoziiert sind, was auf gemeinsame genetische Wege hinweist, die das Risiko für HF beeinflussen.
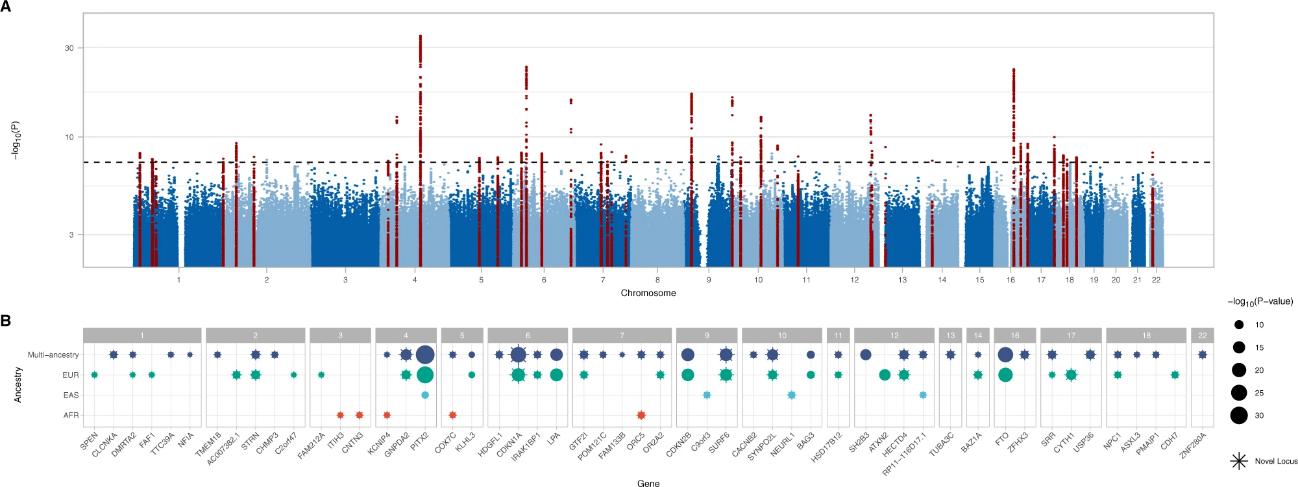 Abb. 1: Genomweite Assoziationen für Herzinsuffizienz.
Abb. 1: Genomweite Assoziationen für Herzinsuffizienz.
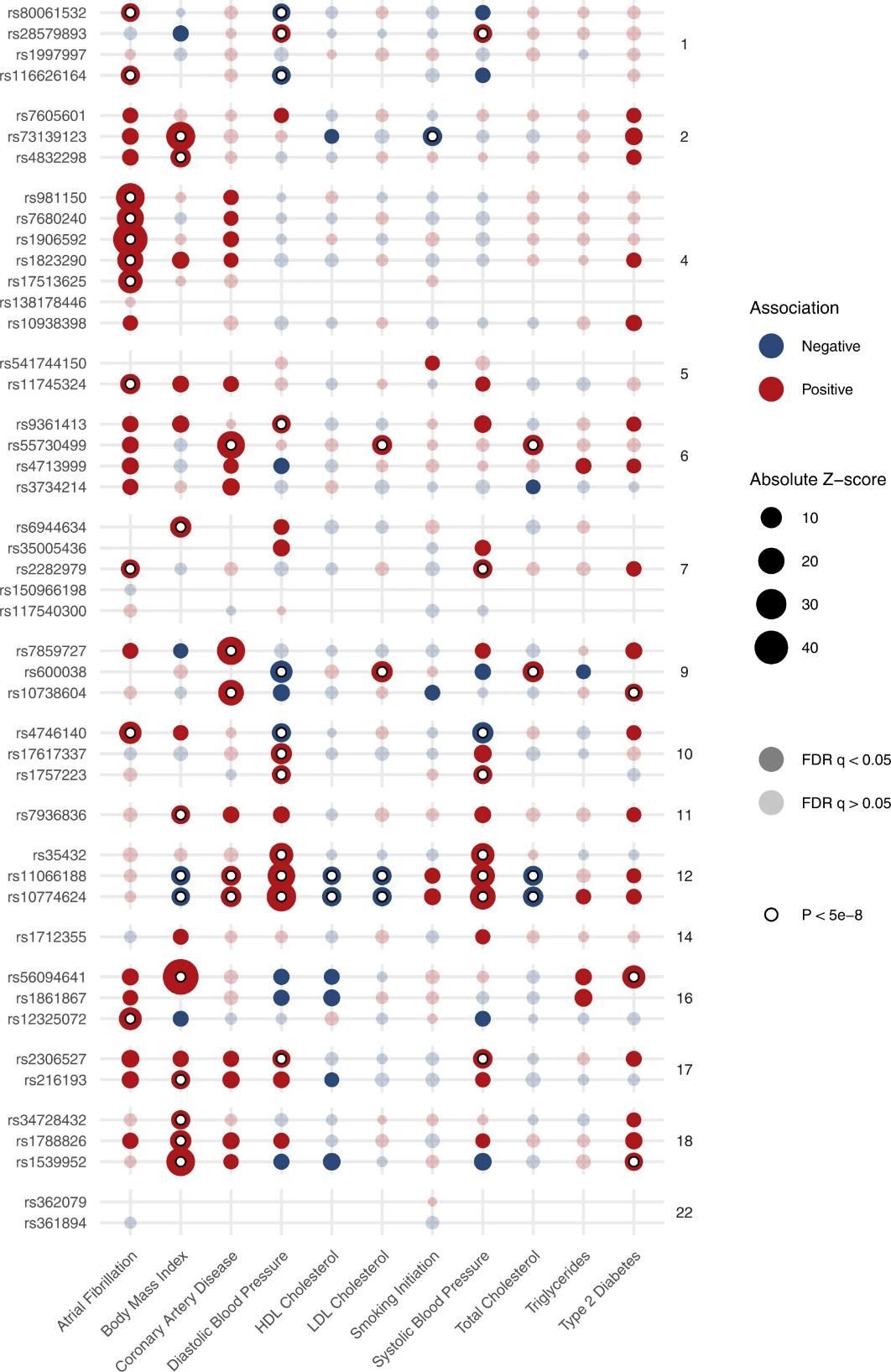 Abb. 2: Assoziationen von Risikovarienten für Herzinsuffizienz mit häufigen kardiometabolischen Merkmalen.
Abb. 2: Assoziationen von Risikovarienten für Herzinsuffizienz mit häufigen kardiometabolischen Merkmalen.
Durch den Einsatz multivariater genetischer Analysemethoden (wie N-GWAMA, MTAG und Genomic Structural Equation Modeling) identifizierten die Forscher 61 unabhängige Loci, die mit Herzinsuffizienz (HF) und verwandten kardiologischen Bildgebungsphänotypen assoziiert sind. Darunter waren 14 neuartige Entdeckungen. Viele dieser Loci sind in der Nähe bekannter Kardiomyopathie-Gene angereichert, was auf eine gemeinsame genetische Ätiologie mit HF und kardiologischen Bildgebungsmerkmalen hinweist. Eine Koinzidenzanalyse dieser Loci über mehrere Merkmale hinweg deutete auf gemeinsame genetische Ursachen hin. Neuartige Assoziationen wurden auch mit anderen kardiovaskulären Merkmalen und Risikofaktoren für HF in Verbindung gebracht, was die genetische Überlappung und Komplexität von HF hervorhebt.
 Abb. 3: Ergebnisse der multivariaten genomweiten Assoziationsstudie.
Abb. 3: Ergebnisse der multivariaten genomweiten Assoziationsstudie.
Fazit
Diese Studie verwendete multi-ancestrale und multi-trait genomweite Analysen, um neue genetische Varianten zu identifizieren, die mit Herzinsuffizienz (HF) und verwandten kardialen Merkmalen verbunden sind. Es wurde festgestellt, dass die Integration verschiedener Arten von genetischen Daten die Entdeckung von HF-Loci verbesserte, wichtige Gene und Wege, die an HF beteiligt sind, hervorhob und potenzielle Verbindungen zwischen zirkulierenden Metaboliten und kardialen Merkmalen aufdeckte. Die Ergebnisse betonen den Wert der Kombination verschiedener genetischer Analysen, um HF und ihre zugrunde liegenden Mechanismen besser zu verstehen.
Referenz
- Levin MG, Tsao NL, Singhal P, et al. Genomweite Assoziations- und Multi-Trait-Analysen charakterisieren die gemeinsame genetische Architektur von Herzinsuffizienz. Naturkommunikation2022, 14. Nov.; 13(1):6914.
Verwandte Veröffentlichungen
Hier sind einige Veröffentlichungen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Sammlung genetischer Daten in ethnisch basierten Studien bei Aymaras, Quechuas und Mestizen: die Herausforderungen der Genetik von Alzheimer in der peruanischen Bevölkerung (GAPP) Studie
Zeitschrift: Alzheimer & Demenz
Jahr: 2022
Bewertung von Plasma-Biomarkern für die A/T/N-Klassifikation der Alzheimer-Krankheit bei Erwachsenen karibischer hispanischer Ethnie
Journal: JAMA Netzwerk Open
Jahr: 2023
Erhöhte Produktion von pathogenen, luftgetragenen Pilzsporen bei der Exposition einer Bodenmykobiota gegenüber chlorierten aromatischen Kohlenwasserstoffschadstoffen.
Journal: Mikrobiologie-Spektrum
Jahr: 2023
Eine Spleißvariante im SLC16A8-Gen führt zu einem Defizit im Laktattransport in aus menschlichen iPS-Zellen abgeleiteten retinalen Pigmentepithelzellen.
Zeitschrift: Zellen
Jahr: 2021
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


