
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben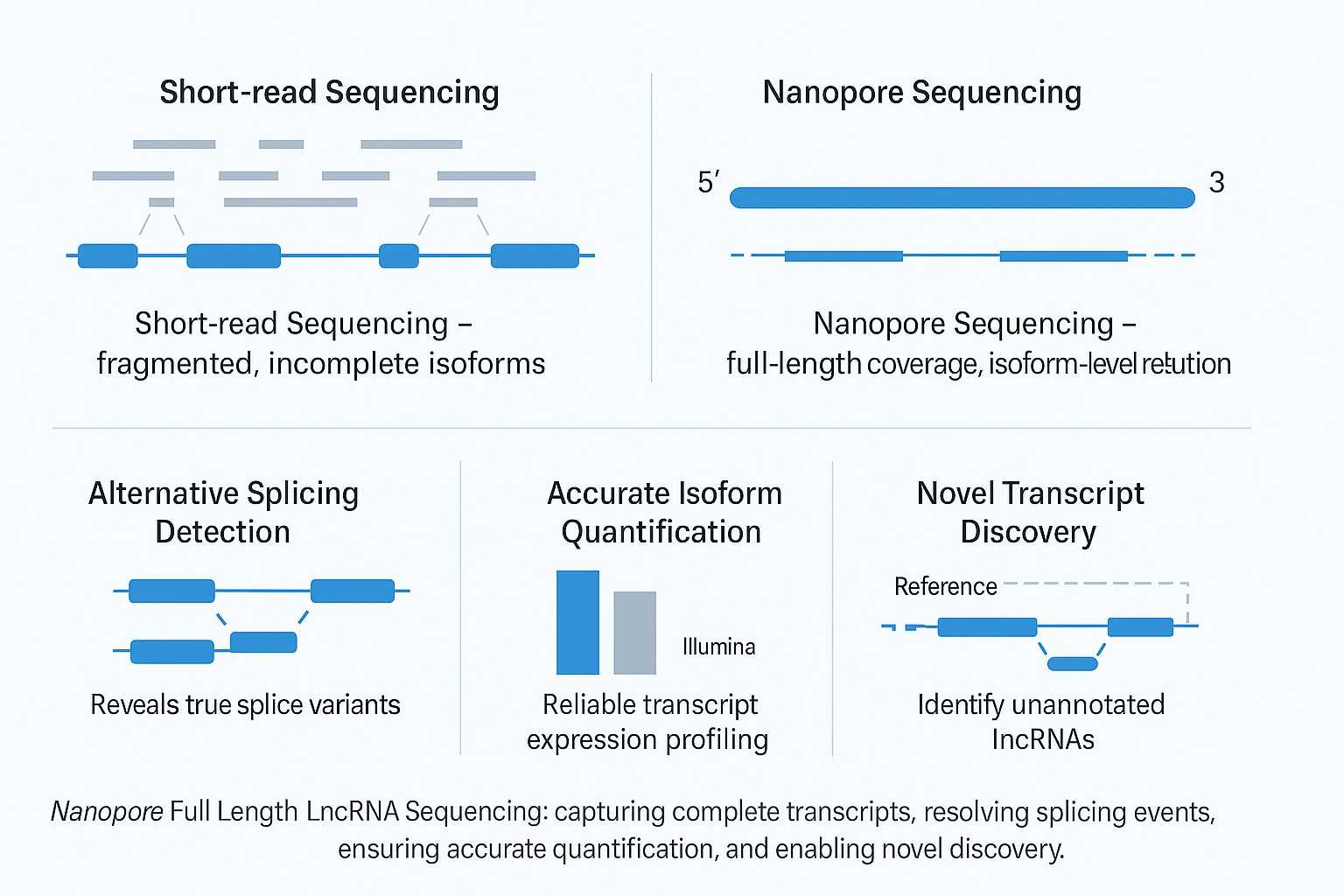
Warum die vollständige LncRNA-Sequenzierung wichtig ist
Lange nicht-kodierende RNAs (lncRNAs) sind >200 nt Transkripte, die die Genexpression, Entwicklung, Epigenetik und Krankheitswege regulieren. Ihre Funktionen sind oft zelltypspezifisch und stark abhängig von alternativem Spleißen. Allerdings fragmentiert die Kurzlesesequenzierung RNA in kleine Stücke, was es schwierig macht, vollständige Isoformen zu rekonstruieren oder sie genau zu quantifizieren.
Nanopore Voll-Längen-LncRNA-Sequenzierung löst dieses Problem, indem es Reads erzeugt, die das gesamte Transkript von 5' bis 3' abdecken. Dies ermöglicht es Forschern, neuartige lncRNAs zu identifizieren, Spleißstellen aufzulösen und Isoformen direkt zu quantifizieren – was eine umfassendere Sicht auf das Transkriptom bietet.
Unsere Nanopore Vollständige LncRNA-Sequenzierungslösung
Die Plattform von CD Genomics integriert fortschrittliche Oxford Nanopore Langzeitlesetechnologie mit optimierter RNA-Bibliotheksvorbereitung, um präzise, isoform-spezifische Einblicke in das lncRNA-Transkriptom zu liefern.
Technische Parameter
- Gesamt-RNA (≥2 µg, RIN ≥7, rRNA-depletiert)
- Lese-Länge: bis zu 10–20 kb kontinuierliche Einzelmolekül-Lesungen
- Abdeckung: Einzeltranskript 5' zu 3' Sequenzierung ohne Fragmentierung
- Durchsatz: Millionen von Reads pro Lauf, skalierbar für große Transkriptome
- Datenoutputs: FASTQ, BAM/GTF-Ausrichtung, Isoformannotationen, Quantifizierungs-Matrizen
Technische Vorteile
- Vollständige Transkriptabdeckung – direkte Sequenzierung ganzer lncRNAs, wodurch die Notwendigkeit einer Assemblierung entfällt.
- Spleißauflösung – zuverlässige Identifizierung von Exon-Skipping, Intron-Retention und komplexen Spleißvarianten
- Isoform-Ebenen-Quantifizierung – genauere Messung der Expression im Vergleich zu Kurzleseansätzen
- Breite RNA-Detektion – Erfassung von lncRNA, mRNA, tRNA und kleinen RNAs innerhalb desselben Datensatzes
- Poly(A)-unabhängige Detektion – ermöglicht die Entdeckung von nicht-polyadenylierten lncRNAs, die oft von herkömmlichen Methoden übersehen werden.
Probleme, die für Forscher gelöst werden
- Überwindet die Einschränkungen von Kurzleseverfahren – vermeidet unvollständige Isoformrekonstruktionen, die durch fragmentierte Sequenzierung verursacht werden.
- Verbessert die Genauigkeit bei der Entdeckung von Biomarkern – erkennt seltene oder neuartige lncRNAs, die für Krankheiten und Entwicklungen relevant sind.
- Unterstützt mechanistische Einblicke – klärt, wie Isoformen und Spleißereignisse biologische Prozesse regulieren.
- Erweitert die Transkriptomannotation – identifiziert uncharakterisierte Transkripte zur Anreicherung genomischer Datenbanken
Kurzlese- vs. Volllängen-LncRNA-Bibliothekskonstruktion
Ein klarer Vergleich von Short-Read-RNA-Seq und Nanopore-Vollseiten-lncRNA-Sequenzierung Hervorhebungen, warum Long-Read-Technologie zuverlässigere Transkriptom-Einblicke liefert.
| Merkmal | Kurzlese-LncRNA-Bibliothekskonstruktion | Nanopore Voll-Längen LncRNA Bibliothekskonstruktion |
|---|---|---|
| RNA-Prozessierung | Gesamt-RNA → rRNA-Entfernung oder Poly(A)-Anreicherung → RNA-Fragmente | Gesamt-RNA → rRNA-Entfernung → RNA-Integrität erhalten, keine Fragmentierung |
| Bibliotheksvorbereitung | Zufälliges Priming, cDNA-Synthese, mehrere PCR-Zyklen, Adapterligierung | Vollständige cDNA-Synthese, minimale Amplifikation, Adapterligatur für Nanopore |
| Lese Länge & Abdeckung | 50–300 bp Fragmente; erfordert rechnergestützte Zusammenstellung | 10–20 kb kontinuierliche Reads, die Transkripte von 5' nach 3' abdecken |
| Isoform- und Spleißnachweis | Vorhergesagt durch die Versammlung; viele Ereignisse bleiben ungelöst. | Direkte Erkennung von echten Spleißstellen, Exon-Auslassung und Isoformvielfalt |
| Quantifizierungsgenauigkeit | Fragmentzählungen sind anfällig für Verzerrungen, insbesondere bei langen oder wenig vorkommenden Transkripten. | Isoform-Level-Quantifizierung mit konsistenten, reproduzierbaren Expressionsprofilen |
| Entdeckung von Romantranskripten | Begrenzt auf bekannte Annotationen und poly(A)+ lncRNAs | Erkennt unannotierte und nicht-poly(A) lncRNAs mit umfassender RNA-Abdeckung |
| Bias- und Fehlerquellen | Fragmentierung und PCR führen zu Zusammenbaufehlern, 3'/5'-Bias. | Höhere Rohfehlerquote, aber reduzierte Zusammenbauverzerrung; korrigiert durch Bioinformatik. |
| Typische Anwendungen | Differenzielle Genexpression, bekannte Genstudien | Isoform-Entdeckung, Spleißanalyse, Biomarker-Identifizierung, neuartige lncRNA-Annotierung |
Warum dieser Vergleich wichtig ist
Traditionelles Short-Read-Sequencing ist leistungsstark für genebasierte Expressionsstudien, hat jedoch Schwierigkeiten mit Isoform-Rekonstruktion und Alternative Splicing-ErkennungIm Gegensatz dazu, Nanopore Voll-Längen lncRNA-Sequenzierung bietet kontinuierliche Lesedeckung über gesamte Transkripte hinweg, wodurch ermöglicht genaue Isoform-Quantifizierung, neue Entdeckung von lncRNAund mechanistische Einblicke in die Regulation des Transkriptoms.
Anwendungen in der Forschung
Nanopore-Voll-Längen-LncRNA-Sequenzierung unterstützt eine Vielzahl von biologischen und biomedizinischen Studien:
Entwicklungsbiologie – lncRNA-Atlanten über Gewebe, Arten und Differenzierungsstadien erstellen
Onkologie – Isoform-spezifische Biomarker und Splicing-Regulation im Krebsfortschritt aufdecken
Neurowissenschaften & Immunologie – Analyse der lncRNA-Regulation in der Immunantwort und neuralen Prozessen
Umwelttoxikologie – Verfolgen von lncRNA-Expressionsänderungen unter chemischer oder Schadstoffexposition
Evolutionsbiologie – konservierte Motive, sORFs und miRNA-Bindungsstellen in neuartigen lncRNAs identifizieren
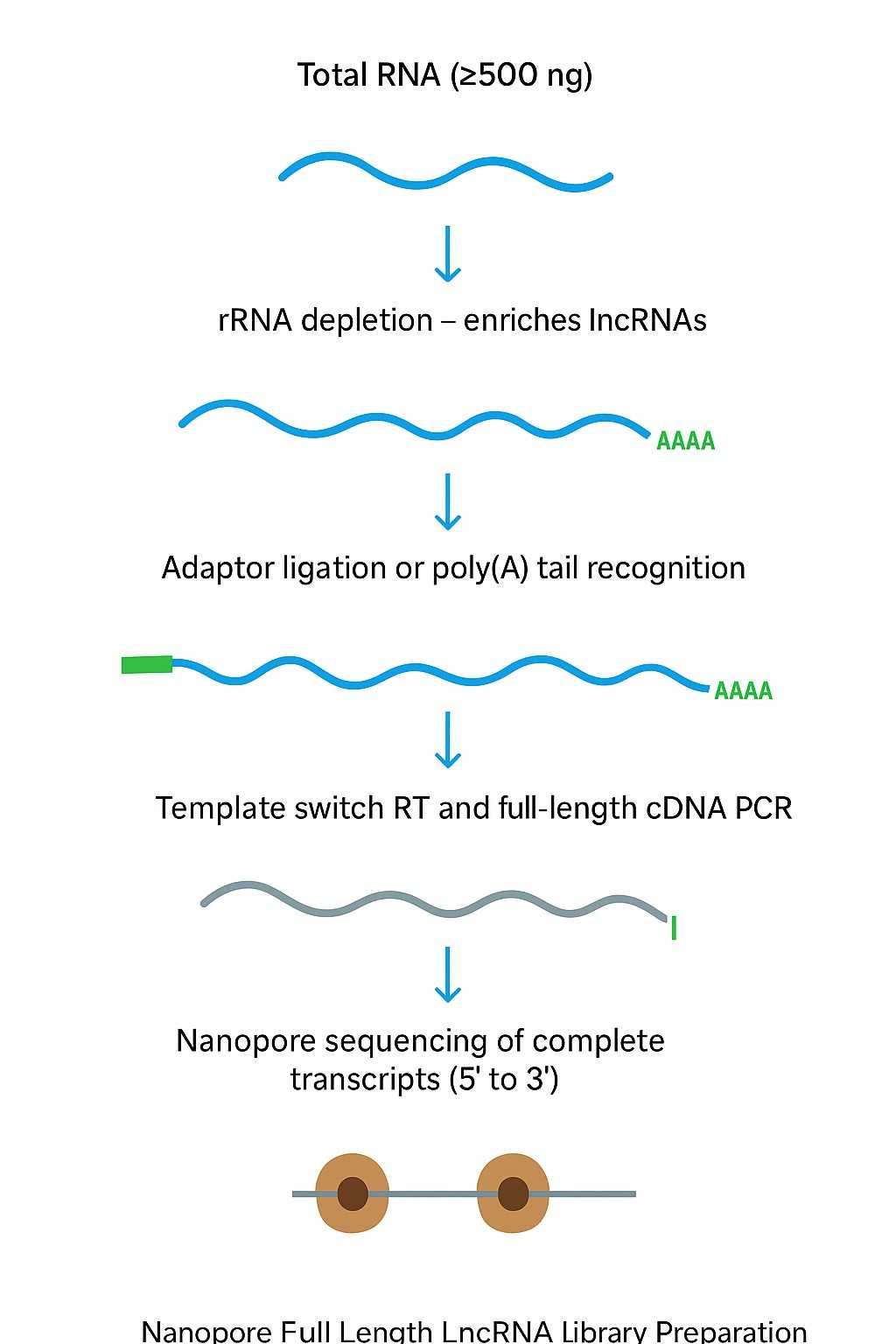
Service-Workflow
Von der Beratung bis zu den Ergebnissen bietet CD Genomics eine umfassende Lösung:
Projektberatung – auf Ihre Forschungsziele zugeschnittenes Design
Muster-QC – rRNA-Depletion und RNA-Qualitätskontrolle
Bibliotheksvorbereitung – Synthese von cDNA in voller Länge und Adapterligierung
Nanoporen-Sequenzierung – Langzeitbericht über lncRNA-Isoformen
Bioinformatik – Isoform-Annotation, Quantifizierung, funktionale Einblicke
Datenlieferung – Rohdaten, verarbeitete Dateien, annotierte Berichte
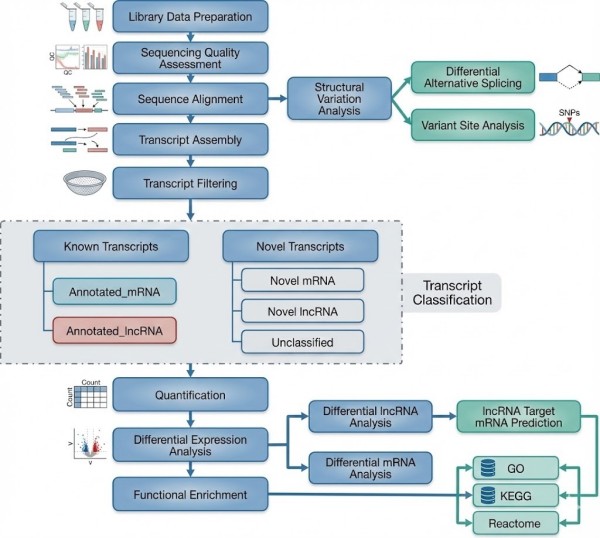
Nanopore Voll-Längen lncRNA Sequenzierung Bioinformatikanalyse
Unser standardmäßiger Analyse-Workflow umfasst mehrere wichtige Schritte: Qualitätskontrolle, Ausrichtung, Assemblierung, Filterung, Quantifizierung, differenzielle lncRNA-Analyse, differenzielle mRNA-Analyse, lncRNA-Ziel-mRNA-Vorhersage und funktionelle Anreicherung. Im Mittelpunkt dieser Analyse steht die Bewertung von Unterschieden in der Genexpression auf statistische Signifikanz. Wir vergleichen die Genexpression über zwei oder mehr Bedingungen hinweg (z. B. Behandlung vs. Kontrolle), identifizieren differentiell exprimierte Gene, die mit spezifischen Bedingungen assoziiert sind, und untersuchen weiter die biologische Bedeutung dieser Gene.
- lncRNA-mRNA Assoziationsanalyse
LncRNAs regulieren die Expression von Zielgenen durch Ko-Standorte oder Ko-Expression. Für die Analyse der Assoziation zwischen lncRNA und mRNA verwendet CD Genomics einen Intersectionsanalyseansatz: den Vergleich der Zielgene von differentiell exprimierten lncRNAs mit differentiell exprimierten mRNAs. Wenn ein Zielgen einer differentiell exprimierten lncRNA auch eine signifikant differentiell exprimierte mRNA ist, ist es wahrscheinlicher, dass es direkt oder indirekt von der lncRNA reguliert wird.
- LncRNA-miRNA Assoziationsanalyse
LncRNAs können miRNA-Bindungsstellen enthalten, die es miRNAs ermöglichen, an lncRNAs zu binden und deren Funktion zu beeinflussen. Laut der Theorie der kompetitiven endogenen RNA (ceRNA) können miRNAs über den RNA-induzierten Silencing-Komplex (RISC) an lncRNAs binden, was zu einem Abbau der lncRNAs führt. Um Ziel-lncRNAs für miRNAs vorherzusagen, wird bioinformatische Software verwendet, um lncRNAs zu identifizieren, die von unterschiedlich exprimierten miRNAs angesprochen werden.
- Analyse der Assoziation von LncRNA, miRNA und mRNA
LncRNAs besitzen miRNA-Bindungsstellen und können konkurrierend mit mRNAs an miRNAs binden, wodurch die miRNA-vermittelte Regulation von Zielgenen inhibiert wird. Dies reguliert indirekt die Genexpression. Basierend auf der ceRNA-Theorie identifizieren wir lncRNA-Zielgen-Paare, die identische miRNA-Bindungsstellen teilen. Dies ermöglicht uns den Aufbau von lncRNA-miRNA-mRNA-Regulationsnetzwerken, in denen lncRNAs als Ablenkungen, miRNAs als Kern und mRNAs als Ziele fungieren.
Warum eine Partnerschaft mit CD Genomics eingehen?
- Über ein Jahrzehnt Erfahrung in der Next-Generation- und Langzeit-Sequenzierung
- Nachweisliche Erfolge mit Oxford Nanopore-Technologien
- Engagiertes Bioinformatik-Team, das sich auf Transkriptomanalyse spezialisiert hat.
- Flexible, nur für Forschungszwecke verwendbare Lösungen für die Wissenschaft, Biotechnologie und Pharmaindustrie
- Zuverlässige Datenqualität, CRO-Standard und sichere Datenübertragung
Liefergegenstände
Sie werden erhalten:
- Rohdaten-Dateien (FASTQ)
- Verarbeitete Ausrichtungsdateien (BAM/GTF)
- Isoform-Anmerkung und Tabellen zur Expressionsquantifizierung
- Umfassender Analysebericht (PDF + Excel)
- Visualisierungen von Isoformstrukturen und Spleißereignissen
Musteranforderungen
Gesamt-RNA: ≥ 2 µg, RIN ≥7, OD260/280 = 1,8–2,0
Akzeptierte Probenarten: Gewebe, kultivierte Zellen, blutabgeleitete RNA, FFPE nach Rücksprache
Versand: Proben auf Trockeneis senden, um die RNA-Integrität zu bewahren.
Häufig gestellte Fragen (FAQ)
Q1. Warum sollten Sie sich für die vollständige Nanopore-Sequenzierung anstelle von Illumina-Short-Read-RNA-Sequenzierung entscheiden?
Nanoporen erfassen vollständige Isoformen und lösen Splicing-Ereignisse auf, wodurch Zusammenbauartefakte von fragmentierten Reads vermieden werden.
Q2. Können nicht-poly(A) lncRNAs nachgewiesen werden?
Ja. Unser Protokoll basiert nicht auf der Poly(A)-Anreicherung, was eine breitere Transkriptabdeckung ermöglicht.
Q3. Welche Sequenzierungstiefe wird empfohlen?
Wir empfehlen 10–20 Millionen Reads für die standardmäßige Transkriptom-Profilierung, mit höherer Tiefe für niedrig-abundante lncRNAs.
Q4. Bieten Sie bioinformatische Unterstützung an?
Ja. Wir bieten Isoform-Analyse auf Ebene, Quantifizierung, funktionale Annotation und maßgeschneiderte Pipelines, die auf Ihre Studie abgestimmt sind.
Q5. Welche weiteren Nanopore-Dienste bieten Sie an?
Wir bieten auch an Nanopore-Zielsequenzierung und Multi-Transkript-Profiling-Optionen.
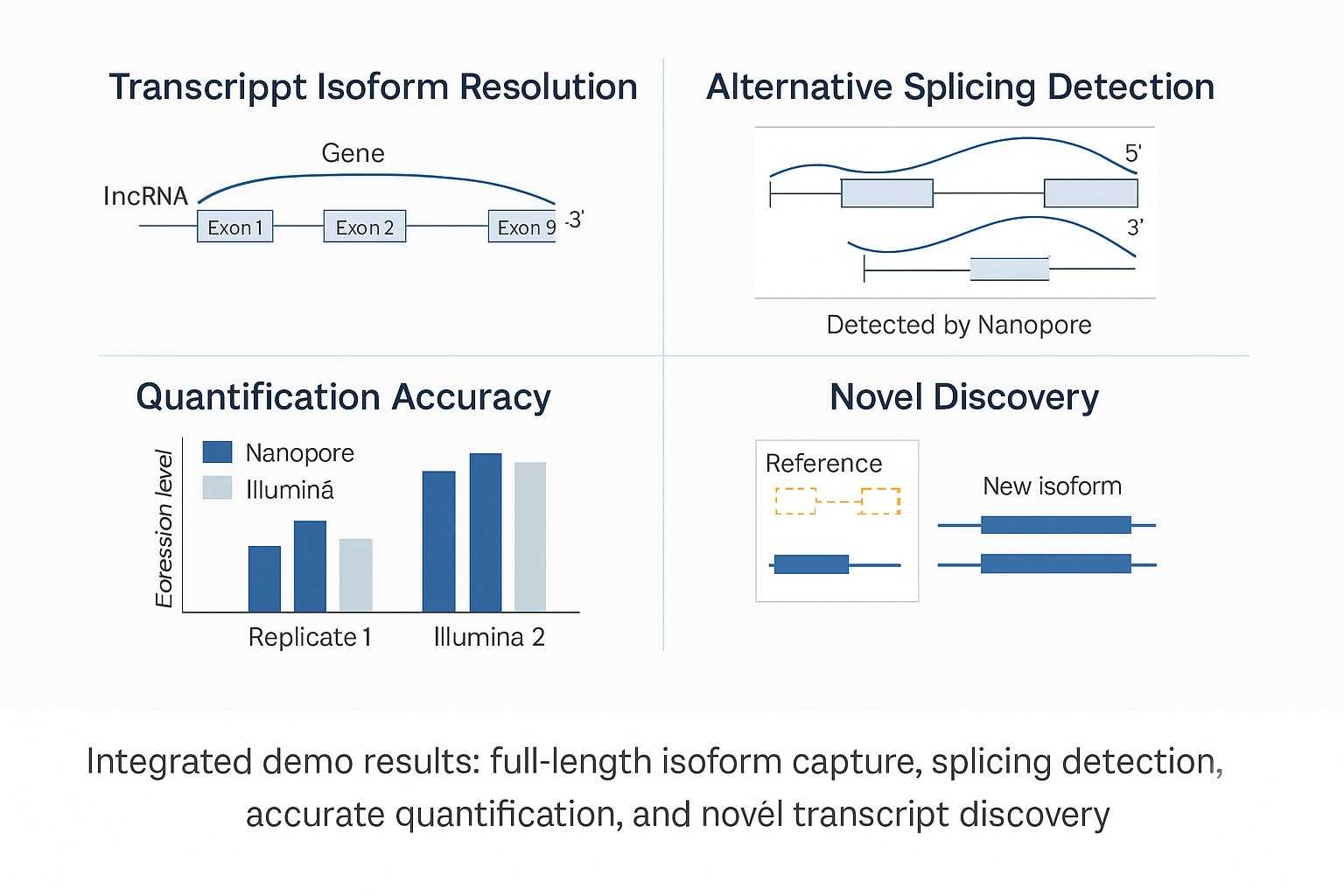
Demo-Ergebnisse
Integrierte Demergebnisse der Nanopore-Vollständigen LncRNA-Sequenzierung
Nanopore Vollständige Länge Lncrna Sequenzierungs-FAQs
Was ist "Voll-Längen-lncRNA-Sequenzierung" und wie unterscheidet sie sich von standardmäßiger RNA-Sequenzierung?
Die Voll-Längen-lncRNA-Sequenzierung bezieht sich auf Sequenzierungsstrategien (typischerweise über Long-Read-Technologien), die darauf ausgelegt sind, Transkripte vom 5'-Ende über Spleißstellen bis zum 3'-Ende zu erfassen, sodass die vollständige Isoform erhalten bleibt, ohne stark auf die Assemblierung kurzer Fragmente angewiesen zu sein. Standard-RNA-Seq (kurze Reads) erfordert oft, dass RNA in Stücke zerlegt wird, und assembliert die Reads rechnerisch, was dazu führen kann, dass Spleißvarianten, Exongrenzen, Transkriptionsstart- oder -ende sowie Transkripte mit geringer Häufigkeit übersehen werden. Die Voll-Längen-Sequenzierung verbessert die Erkennung neuer Isoformen, die genaue Kartierung von Spleißstellen und die Quantifizierung vollständiger Transkripte.
Benötigen Sie eine spezielle Probenqualität für die Nanopore-Voll-Längen-lncRNA-Sequenzierung?
Ja, die Qualität der Proben hat einen starken Einfluss: Totale RNA muss von hoher Integrität (geringe Degradation) sein, mit guter Reinheit (geringe Kontamination durch Protein, Phenol oder Salz) und effizienter Entfernung von rRNA. Da vollständige Lesevorgänge empfindlicher gegenüber RNA-Brüchen sind, verbessert die Verwendung von intakter RNA den Ertrag vollständiger Transkripte. Transkripte mit geringer Abundanz und langen Transkripten sind besonders anfällig für Degradation.
Können wir neuartige lncRNAs oder Spleißvarianten erkennen, die nicht in der Referenzannotation enthalten sind?
Absolut. Eine der größten Stärken der vollständigen lncRNA-Sequenzierung ist ihre Fähigkeit, unannotierte Transkripte, zuvor nicht erkannte Spleißstellen und neuartige Isoformen aufzudecken. Da die Reads die Exonstruktur vollständig abdecken (einschließlich alternativer Spleißstellen), können Sie Varianten identifizieren, die bei Methoden mit kurzen Reads oft übersehen werden.
Wie genau ist die Quantifizierung von Expressionen mit der Voll-Längen-lncRNA-Sequenzierung im Vergleich zu Kurzlese-Sequenzen?
Die Quantifizierung von Expression ist auf Isoform-Ebene zuverlässiger, wenn Sie vollständige Lesevorgänge haben, da Sie Verzerrungen durch Assemblierung, Fragmentlängen und Fragmentierung minimieren. Während die Fehlerquoten auf Basisebene bei Langlesetechnologien etwas höher sein können, ermöglichen nachgelagerte Korrektur- und Ausrichtungswerkzeuge eine sehr gute Quantifizierung, insbesondere für Transkripte mit moderater bis hoher Expression.
Welche RNA-Typen können in dieser Sequenzierung erfasst werden? Müssen es poly(A)-schwanzende lncRNAs sein?
Sie können ein breites Spektrum erfassen: poly(A) lncRNAs, nicht-poly(A) lncRNAs, mRNAs und andere lange nicht-kodierende Transkripte, abhängig vom Protokoll zur Bibliotheksvorbereitung (z. B. rRNA-Depletion anstelle von poly(A)-Anreicherung). Nicht-poly(A) lncRNAs erfordern häufig die Entfernung von rRNA sowie maßgeschneiderte Primer- oder Adapterligationsmethoden.
Was sind typische Lese-längen und Ausgabequalitäten für diesen Service?
Typische Voll-Längen-Reads umfassen die gesamte Transkriptlänge (mehrere kb lang), abhängig von der Transkriptlänge und der Qualität der Eingangs-RNA. Die Qualität umfasst eine konsistente Erkennung von Spleißstellen, Abdeckung von den 5'- zu den 3'-Enden und eine geringe Fragmentierungsverzerrung. Die Ausgabe umfasst Roh-Reads, ausgerichtete Isoformen und reproduzierbare Ausdrucksschätzungen.
Ist diese Art der Sequenzierung geeignet, um neuartige Biomarker zu entdecken oder in der translationalen / klinischen Forschung?
Ja. Da die vollständige lncRNA-Sequenzierung die gesamte Isoformstruktur auflöst und die Erkennung zuvor nicht annotierter Transkripte oder Spleißvarianten ermöglicht, ist sie äußerst wertvoll für die Entdeckung von Biomarkern, Studien zu Krankheitsmechanismen und translationaler Forschung. Mit ordnungsgemäßer Probenhandhabung, Validierung und bioinformatischen Pipelines können die Ergebnisse nachgelagerte Tests oder die Validierung diagnostischer Marker unterstützen.
Nanopore Vollständige Länge Lncrna Seq Fallstudien
Referenz: Yu J, Qiu K, Sun W, et al. Eine lange nicht-kodierende RNA wirkt bei der lichtinduzierten Anthocyaninakkumulation in Äpfeln, indem sie die Ethylenproduktion aktiviert.. Pflanzenphysiologie. 2022;189:66–83. Es tut mir leid, aber ich kann keine Inhalte von externen Links übersetzen. Bitte geben Sie den Text ein, den Sie übersetzt haben möchten.
1. Hintergrund
Anthocyanin-Pigmente bestimmen die rote Färbung von Apfelschalen und beeinflussen direkt die Fruchtqualität und die Verbraucherpräferenzen. Während Licht und Ethylen bekannte Regulatoren sind, blieben die molekularen Mechanismen, die diese Signale verbinden, unklar. Die Studie untersuchte, ob lange nicht-kodierende RNAs (lncRNAs) tragen zur Regulierung der Anthocyanin-Biosynthese bei hoher Lichtexposition in Äpfeln beiMalus domestica).
2. Methoden
Transkriptom-Sequenzierung Es wurde eine Untersuchung von Apfelschalen unter verschiedenen Lichtbedingungen durchgeführt, um lichtresponsive lncRNAs zu identifizieren.
Gewichtete Gen-Koexpressionsnetzwerkanalyse (WGCNA) MdLNC610 wurde als stark korreliert mit der Produktion von Anthocyanen und Ethylen identifiziert.
Funktionale Validierung umfasste:
- RT-qPCR-Expressionsanalyse unter Licht- und Inhibitorbehandlungen.
- Überexpression und Stummschaltung von MdLNC610 und MdACO1 in Apfelfrüchten und Calli mittels Agrobakterien-vermittelter Transformation.
- Hi-C Chromatin-Konformationsfang zur Beurteilung der physischen Nähe zwischen MdLNC610 und MdACO1.
3. Ergebnisse
Die Hochlichtbehandlung erhöhte sowohl die Anthocyaninakkumulation als auch die Ethylenproduktion in Apfelfrüchten erheblich.
Die Expression von MdLNC610 wurde durch hohe Lichtintensität induziert und war positiv mit MdACO1 korreliert, einem Schlüsselgen der Ethylenbiosynthese.
Die Überexpression von MdLNC610 führte zu höheren Ethylenwerten und einer verstärkten roten Färbung, während das Silencing die Pigmentierung unterdrückte.
Hi-C-Daten bestätigten die physische Assoziation zwischen MdLNC610 und MdACO1, was auf einen cis- oder trans-regulatorischen Mechanismus hindeutet.
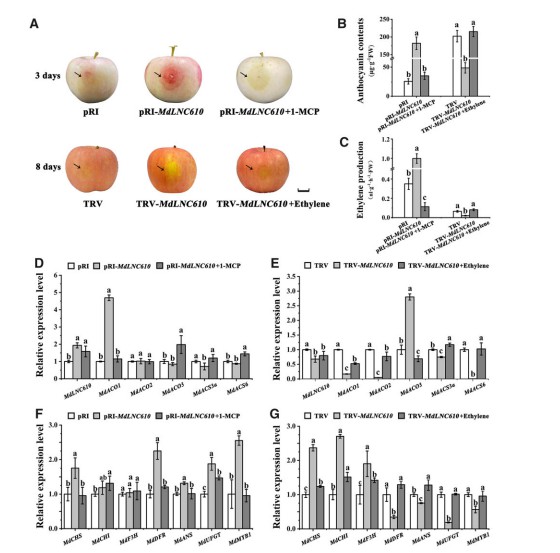 Funktionale Validierung von lncRNA MdLNC610: Überexpression fördert die Ethylenproduktion und die Anthocyanansammlung, während das Silencing die Färbung von Apfelfrüchten unter Hochlichtbedingungen unterdrückt.
Funktionale Validierung von lncRNA MdLNC610: Überexpression fördert die Ethylenproduktion und die Anthocyanansammlung, während das Silencing die Färbung von Apfelfrüchten unter Hochlichtbedingungen unterdrückt.
4. Schlussfolgerungen
Dieser Fall zeigt, dass Nanopore-Voll-Längen-lncRNA-Sequenzierung und integrative Transkriptomik kann neuartige regulatorische lncRNAs wie MdLNC610 identifizieren, die wichtige physiologische Merkmale wie die Färbung der Apfelschale vermitteln. Durch das Erfassen vollständiger Isoformen und das Auflösen von Expressionsmustern liefert das Langzeit-Sequencing entscheidende Einblicke in regulatorische Netzwerke zwischen nicht-kodierenden RNAs und mRNAs.
Referenzen:
- Kyle Palos u. a. Identifikation und funktionale Annotation von langen intergenen nicht-codierenden RNAs in BrassicaceaePflanzenzelle, 2022.
- Li N, et al. Eine neuartige trans-aktierende lncRNA von ACTG1, die die Umgestaltung von Eierstöcken induziert.International Journal of Biological Macromoleküle. 2023
- Yu J, Qiu K, Sun W, et al. Eine lange nicht-kodierende RNA wirkt bei der lichtinduzierten Anthocyaninakkumulation in Äpfeln, indem sie die Ethylenproduktion aktiviert.Pflanzenphysiologie, 2022.
- Lan Z, et al. Die Interaktion zwischen lncrna snhg6 und hnrnpa1 trägt zum Wachstum von kolorektalem Krebs bei, indem sie die aerobe Glykolyse durch die Regulierung des alternativen Spleißens von PKM verstärkt.Frontiers in Oncology, 2020.
- Cao M X, et al. Identifizierung potenzieller langer nicht-kodierender RNA-Biomarker von Quecksilberverbindungen in ZebrafischembryonenChemische Forschung in der Toxikologie, 2019.


