
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben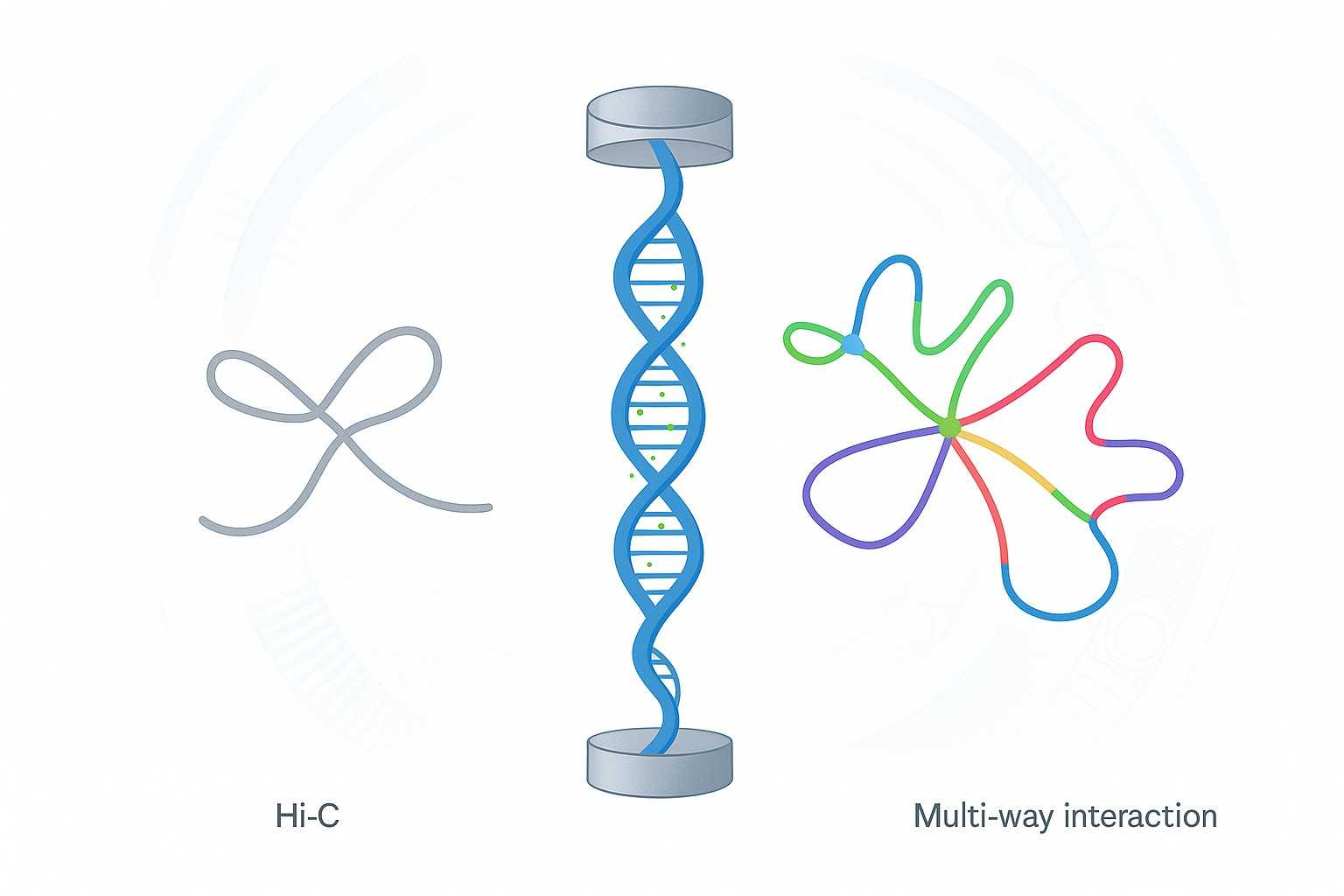
Einführung
Die traditionelle Hi-C-Sequenzierung war das Fundament für das Studium der 3D-Genomarchitektur, aber ihre Beschränkung auf paarweise Wechselwirkungen lässt viele höherordentliche Chromatinstrukturen ungelöst. Mit dem Fortschritt der Genomprojekte zu Telomer-zu-Telomer (T2T)-Assemblierungen, polyploiden Organismen und komplexen Pflanzen- und Tiergenomen stehen die Forscher vor einer wachsenden Herausforderung: Wie können wir Contigs genau verankern, Zentromere auflösen und die wahre Chromatin-Komplexität erfassen?
Pore-C-Sequenzierung, eine auf Nanoporen basierende Methode zur Erfassung der Chromatinstruktur mit langen Reads, bietet die Lösung. Durch die direkte Sequenzierung von ultra-langen Ketten zeigt die Pore-C-Nanoporentechnologie gleichzeitig mehrfache Chromatininteraktionen und DNA-Methylierung, die Einblicke bieten, die über die Reichweite von Hi-C hinausgehen. Für Forscher in biochemischen Laboren, CRO-Zusammenarbeiten und akademischen Institutionen eröffnet Pore-C eine neue Dimension in der Genomassemblierung und epigenetischen Entdeckung.
Technologieübersicht: Was ist Pore-C-Sequenzierung?
Pore-C-Sequenzierung ist fortgeschritten Nanoporen-basierte Chromatin-Konformationsfangtechnologie die die Einschränkungen von Hi-C überwindet. Durch die Kombination von Chromatinvernetzung und Ligation mit Oxford Nanopore Langzeit-SequenzierungPore-C ermöglicht die direkte Erkennung von mehrwegige Chromatin-Interaktionen entlang einzelner DNA-Moleküle.
Wie es funktioniert
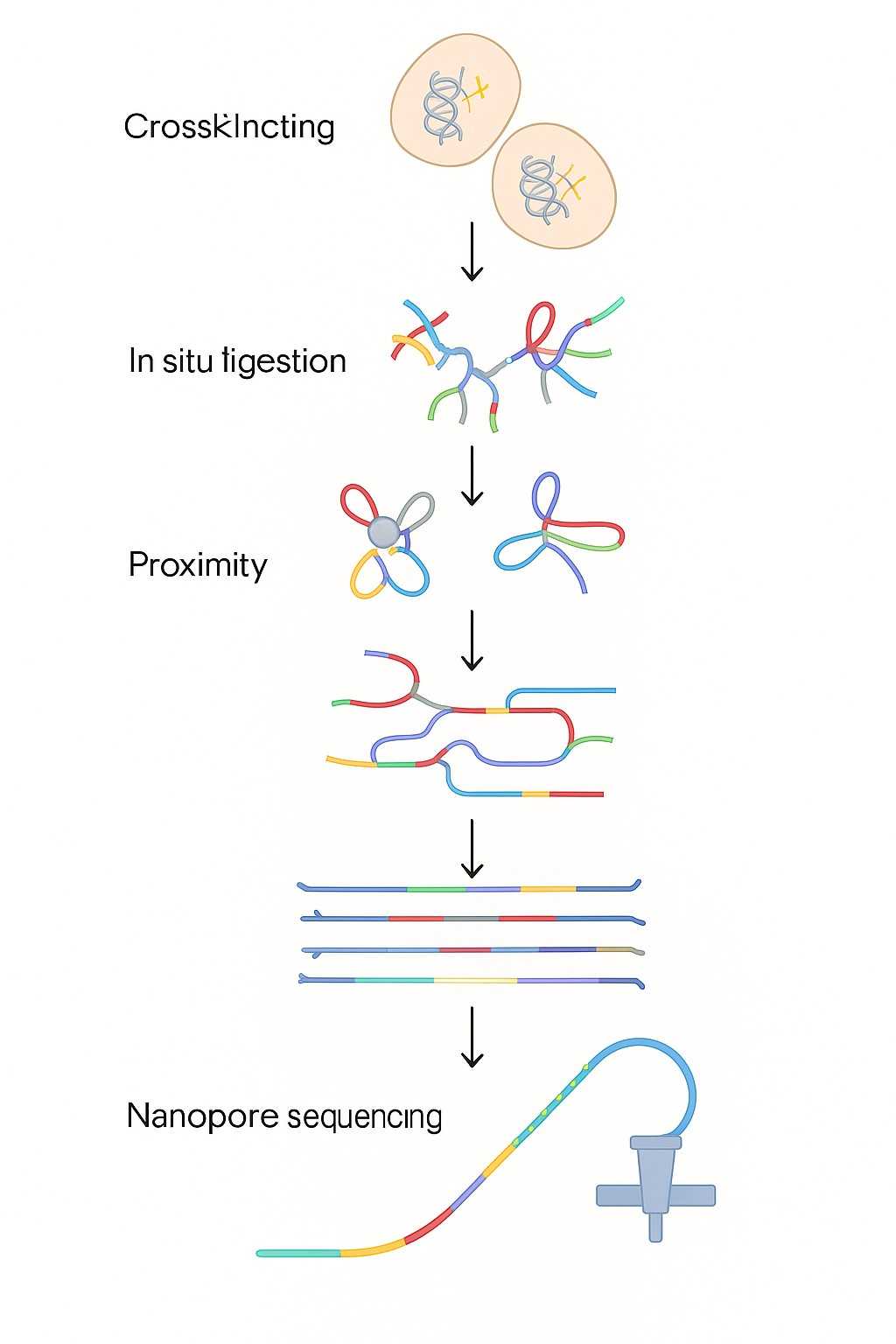
- Kreuzvernetzung & Verdauung: Chromatin-Interaktionen werden durch Formaldehydfixierung und Schneiden mit Restriktionsenzymen erhalten.
- Ligation: DNA-Fragmente, die sich in enger räumlicher Nähe befinden, werden zu Kettenmolekülen ligiert, wobei ihre 3D-Kontaktinformationen erhalten bleiben.
- Nanopore-Sequenzierung: Ultra-lange Kettenmoleküle werden direkt sequenziert, wodurch mehrere interagierende Loci in einem einzigen Lesevorgang erfasst werden.
- Integrierte Daten: Neben 3D-Chromatin-Interaktionen bewahrt die Pore-C-Sequenzierung native DNA-Methylierungssignale und bietet sowohl strukturelle als auch epigenetische Einblicke in einem Arbeitsablauf.
Warum es wichtig ist
- Im Gegensatz zu Hi-C, das auf paarweise Interaktionen beschränkt ist, liefert die Pore-C-Nanoporen-Sequenzierung höherwertige Kontaktkarten, die die wahre Komplexität der Chromatinfaltung widerspiegeln.
- Mehrweg-Interaktionsdaten verbessern die Genauigkeit der Telomer-zu-Telomer-Genomassemblierung, der Zentromerauflösung und der polyploiden Gerüstbildung.
- Die integrierte Methylierungsdetektion fügt der epigenetischen Forschung eine neue Dimension hinzu.
Pore-C vs Hi-C: Eine neue Dimension in der Genomanalyse
| Merkmal | Hi-C | Pore-C |
|---|---|---|
| Interaktionstyp | Nur paarweise (zwei Loci gleichzeitig) | Mehrweg-Interaktionen (3+ Loci in einem einzelnen Lesen) |
| Leseumfang | Short-Read Illumina | Ultra-lange Nanopore-Kontakte |
| Epigenetische Informationen | Nicht erfasst | Direkte DNA-Methylierungsdetektion |
| Genomverankerungstiefe | Erfordert ~100× Abdeckung | Vergleichbare Ergebnisse mit ~30× Abdeckung |
| Komplexe Regionen | Begrenzte Zentromerauflösung | Starke Signale in Zentromeren und Wiederholungen |
| Polyploide Assemblierung | Höheres Risiko von Fehlverknüpfungen zwischen Homologen | Reduzierte falsche Verknüpfungen, höhere Genauigkeit der Gerüste |
| Datenausgabe | Kontakt-Wärmebilder | Kontakt-Wärmekarten + Mehrweg-Netzwerke + Methylierungsspuren |
Warum Pore-C Hi-C übertrifft
- Stärkere Interaktionssignale über das gesamte Genom hinweg, einschließlich der Zentromere.
- Vollständigere Genomassemblierungen sowohl in diploiden als auch in polyploiden Arten.
- Duale Einblicke: 3D-Chromatinstruktur und epigenetische Modifikationen.
- Effiziente Sequenzierungstiefe: 30× Pore-C kann die Ankerkraft von 100× Hi-C erreichen.
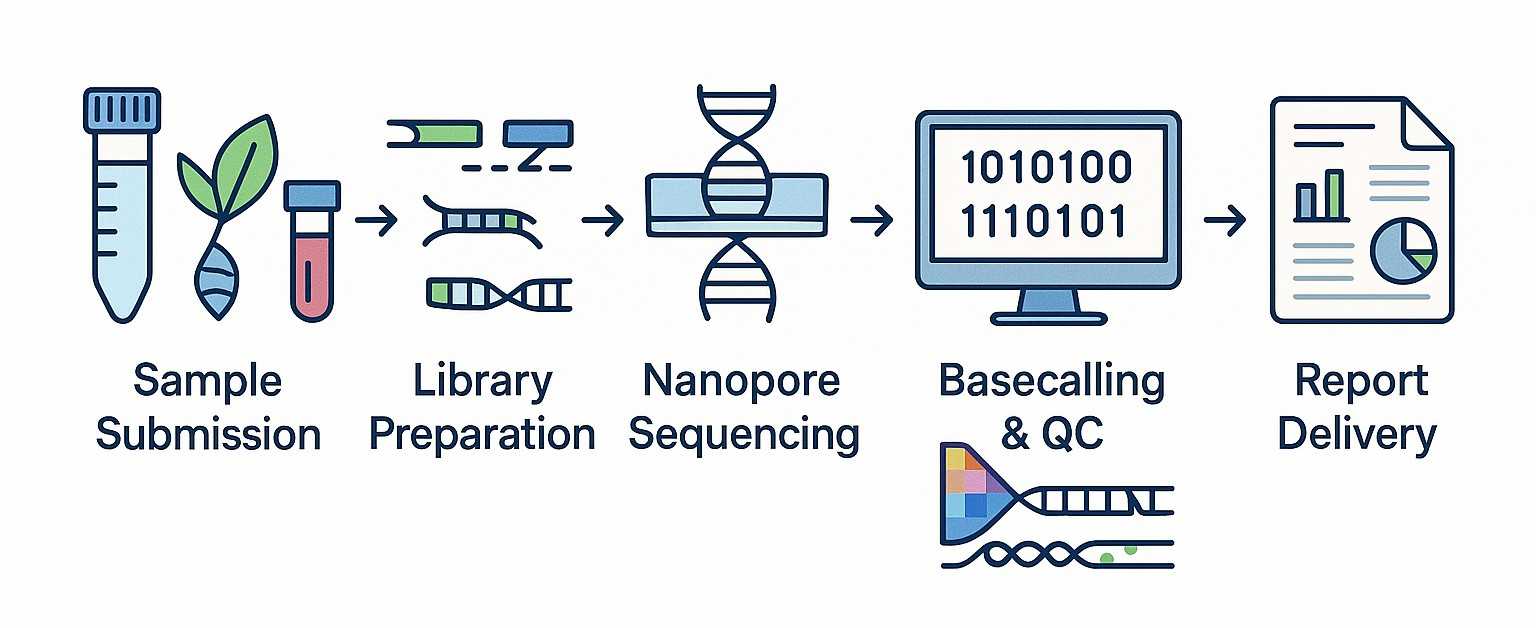
Service-Workflow
Anwendungen der Pore-C-Sequenzierung
Pore-C-Sequenzierung bietet eine leistungsstarke Lösung für Forschungsteams, die über die Grenzen von Hi-C hinausgehen möchten. Mit der Fähigkeit, zu erfassen mehrwegige Chromatin-Interaktionen und erkennen DNA-Methylierung gleichzeitig, Pore-C Nanoporenanalyse unterstützt ein breites Spektrum an Anwendungen in den Lebenswissenschaften und der Landwirtschaft.
Genomassemblierung und Scaffoldierung
- Stärkt die Contig-Verankerung in Telomer-zu-Telomer (T2T) Genomprojekten.
- Verbessert die Auflösung von Zentromeren, repetitiven Regionen und strukturellen Varianten.
- Bietet überlegene Gerüstgenauigkeit für komplexe oder große Genome.
Polyploide Genomforschung
- Reduziert Fehlassemblierungen, indem homologe Chromosomenkreuzsignale minimiert werden.
- Bietet eine klarere Trennung der Subgenome in polyploiden Pflanzen und Tieren.
- Unterstützt eine genaue Haplotype-Phasierung und evolutionäre Studien.
3D-Genomarchitektur
- Ermöglicht die Abbildung höherer chromosomaler Strukturen über paarweise Kontakte hinaus.
- Enthüllt mehrdimensionale Interaktionsnetzwerke, die die Genregulation steuern.
- Fortschritte im grundlegenden Verständnis der nuklearen Organisation.
Epigenetik und Chromatinbiologie
- Erfasst DNA-Methylierungsmuster sowie strukturelle Interaktionen.
- Bietet eine integrierte Sicht auf die epigenetische Regulation im 3D-Raum.
- Ermöglicht Forschung zur Kontrolle der Genexpression, Prägung und Stummschaltung.
Pflanzen- und Tierzucht
- Beschleunigt den Aufbau von Pan-Genomen und Diversitätsstudien.
- Unterstützt die Identifizierung von strukturellen Merkmalen, die mit agronomischen und krankheitsresistenten Eigenschaften verbunden sind.
- Bietet wertvolle Einblicke für Programme zur genetischen Verbesserung.
Empfohlene Pore-C-Strategien für verschiedene Genomebenen
Um die Vorteile von... zu maximieren Pore-C-Sequenzierung Bei der Genomassemblierung werden je nach Zielgenomlevel unterschiedliche Strategien empfohlen. Diese Kombinationen gewährleisten ein optimales Gleichgewicht zwischen Lesegenauigkeit, langfristigen Interaktionen und Sequenzierungstiefe.
| Genom-Ebene | Empfohlene Strategie |
|---|---|
| Chromosomenebene Genom | 30× HiFi + 30× Pore-C + 50× NGS |
| T2T-Genom | 30× HiFi + 30× ONT ultra-lang + 30× Pore-C + 50× NGS |
| Perfektes T2T-Genom | 40–60× HiFi + 60–100× ONT ultra-lang + 30× Pore-C + 50× NGS |
| Haploider perfekter T2T-Genom | 80–120× HiFi + 120–200× ONT ultra-lang + 60× Pore-C + 50× NGS |
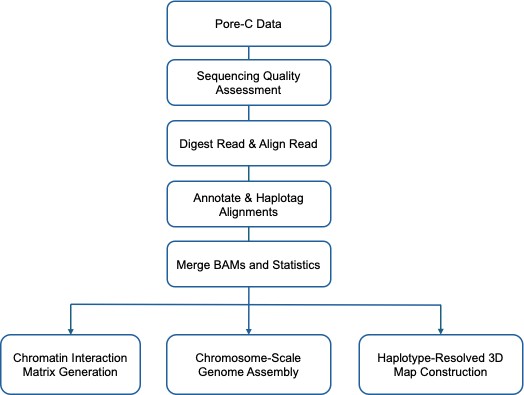
Datenanalyse & Bioinformatik
Der wahre Wert von Pore-C-Sequenzierung liegt nicht nur in der Generierung von Long-Read-Concatemer-Daten, sondern auch in der Extraktion genauer und effizienter Interaktionssignale. Bei CD Genomics setzen wir optimierte Pore-C-Analysepipelines um qualitativ hochwertige Ergebnisse für die Genomassemblierung und die 3D-Genomforschung sicherzustellen.
Optimierte Analyse-Strategie
Traditionelle Hi-C-Pipelines basieren auf einem zuerst schneiden, später ausrichten Methode, die oft die einzigartige Kraft von Pore-C unterschätzt. Stattdessen verwenden wir ein zuerst ausrichten, später schneiden Ansatz, der auf Nanopore-Daten zugeschnitten ist:
- Die vollständige Leseausrichtung an das Referenzgenom bewahrt den langfristigen Kontext.
- Die Fragmentanalyse nach der Ausrichtung vermeidet Überteilungen und reduziert Mehrfachzuordnungsfehler.
- Die genaue Extraktion gültiger Paare erhöht die nutzbaren Datenraten und die Interaktionsauflösung.
Diese Strategie verbessert erheblich die gültige Datenrate im Vergleich zu herkömmlichen Methoden, wodurch sichergestellt wird, dass jeder Pore-C-Lesevorgang effektiver zur nachgelagerten Analyse beiträgt.
Neue QC-Metriken für Pore-C
Über die standardmäßige Generierung von Kontaktkarten hinaus bewertet unser bioinformatischer Workflow Pore-C-Datensätze mit spezialisierten Metriken, einschließlich:
- Mittlere Fragmentanzahl – durchschnittliche Anzahl der Loci, die pro Lesevorgang erfasst werden.
- Kontakte/Lesen-Verhältnis – Effizienz der Interaktionsaufnahme pro Lesevorgang.
- Gültige Größe/Gesamtgröße – Anteil der effektiv genutzten Daten.
- Mittlere gültige Paarlänge – Abstand, der durch gültige Interaktionen überbrückt wird.
Diese Kennzahlen bieten tiefere Einblicke in beide. Datenqualität und biologische Signalstärke, um eine zuverlässige Interpretation zu gewährleisten.
Liefergegenstände
Kunden erhalten ein komplettes Datenpaket, das direkt in Genom- und epigenetische Studien integriert werden kann:
- Rohe Nanopore-Sequenzierungsdaten (FASTQ/FAST5)
- Verarbeitete Interaktionspaare (Mehrwegekontakte)
- Chromatin-Kontaktkarten und 3D-Interaktionsnetzwerke
- DNA-Methylierungsprofile zusammen mit strukturellen Daten
- Umfassender Bioinformatikbericht mit QC-Statistiken und Visualisierungen
Pore-C Datenqualitätsprüfung
Pore-C-Sequenzierung bietet eine hochwertige Datengenerierung mit einem Fokus auf optimale Probenarten für zuverlässige Ergebnisse.
| Der Sequenzierungsindex | Ertrag (/Zelle) | |
|---|---|---|
| Konventionelle Arten | Pass liest N50≥1Kb | Rohdaten ≥50 Gb |
| Musterbeschreibung | 1. Es wird empfohlen, Pflanzenproben aus neu gewachsenen jungen Blättern zu senden. 2. Bei Tiersamples wird empfohlen, frisches Blut den Vorzug zu geben, gefolgt von Leber und anderen Geweben. 3. Unkonventionelle Proben garantieren keinen Sequenzierungs-Ertrag. 4. Nanoporen-Sequenzierung unkonventioneller Proben ist wie folgt: Pflanzen mit vielen sekundären Metaboliten; 2) Ozeane und aquatische Produkte (Tiere und Pflanzen, Algen usw.); 3) Kaninchen, Insekten, Amphibien, Vögel usw. |
|
Beispielanforderungen für Pore-C-Sequenzierung
Um qualitativ hochwertige Ergebnisse zu gewährleisten, befolgen Sie bitte die folgenden Richtlinien beim Vorbereiten und Versenden Ihrer Proben. Alle Proben müssen sein schnellgefroren in flüssigem Stickstoff, gespeichert bei −80 °Cund versandt am Trockeneis um Degradation zu vermeiden.
| Probenart | Empfohlene Menge |
|---|---|
| Zelle | ≥ 106 Zellen |
| Periphere Blutmononukleäre Zellen (PBMCs) | PBMC-Pellet aus ca. 5 ml frischem Vollblut |
| Tiergewebe | ~50-100 mg kryogenisch gemahlenes Gewebe |
| Insektenmaterial | ~50-100 mg kryogenisch gemahlenes Material |
| C. elegans Material | ~1 ml Kryo-geschrotetes Wurm-Pulver |
| Pflanzenmaterial | ~2 g Pflanzenmaterial |
Wichtige Hinweise:
- Kennzeichnen Sie jedes Röhrchen deutlich mit der Proben-ID (Buchstaben + Zahlen) und stellen Sie sicher, dass sie mit dem Probeninformationsformular übereinstimmt.
- Vermeiden Sie Frost-Tau-Zyklen.
- Überschreiten Sie nicht das Fünffache der empfohlenen Eingabemengen, es sei denn, es ist für Proben mit niedrigem Ertrag anders angegeben.
Warum CD Genomics für die Ganzgenomsequenzierung wählen?
Von fortschrittlichen Sequenzierungsplattformen bis hin zu hochwertigen Datenlieferungen bietet CD Genomics eine effiziente, umfassende Lösung, die auf verschiedene Forschungsbedürfnisse zugeschnitten ist. Unser Team gewährleistet zuverlässige Ergebnisse mit flexibler Unterstützung.
- Bewährte FachkompetenzUnser Team verfügt über umfangreiche Erfahrung in Chromatin-Konformations-Techniken, einschließlich Pionierarbeit mit Pore-C seit 2022.
- Optimierte, robuste ProtokolleWir haben jeden Schritt (Kreuzvernetzung, Lyse, Ligation, Reinigung) für maximale Ausbeute und Komplexität verfeinert und dabei Verzerrungen minimiert.
- Hochdurchsatz-SequenzierungDer direkte Zugang zu PromethION 48-Plattformen gewährleistet eine schnelle und umfassende Sequenzierung.
- Fortgeschrittene BioinformatikÜber Standard-Pipelines hinaus bieten wir eine ausgeklügelte Mehrwege-Kontaktanalyse, Integration und maßgeschneiderte Lösungen an.
- Strenge QualitätskontrolleMehrere QC-Prüfpunkte gewährleisten die Datenzuverlässigkeit.
- End-to-End UnterstützungVon der Projektplanung bis zur endgültigen Interpretationsunterstützung.
Pore-C Seq FAQs
Q: Was ist Pore-C und wie unterscheidet es sich von Hi-C?
Pore-C ist eine Methode zur Erfassung der Chromatinstruktur, die anstelle von Kurzlesungen die Langlese-Nanopore-Sequenzierung verwendet; sie erfasst mehrfache (3 oder mehr Loci) Chromatininteraktionen in einzelnen Lesungen, bewahrt die DNA-Methylierung, vereinfacht die Bibliotheksvorbereitung, indem sie Biotinmarkierung und PCR-Schritte eliminiert, und löst komplexe Regionen, die für Hi-C herausfordernd sind.
Kann Pore-C epigenetische Modifikationen und Interaktionen gleichzeitig erkennen?
Ja, weil Pore-C Nanopore-Sequenzierung verwendet, die PCR-frei ist und native DNA-Basenmodifikationen beibehält; somit können sowohl Chromatin-Interaktionsnetzwerke als auch Methylierungssignaturen (zum Beispiel 5mC) aus demselben Datensatz gewonnen werden.
Q: Welche Arten von Proben sind für das Pore-C-Sequencing geeignet?
Proben, die quervernetzte Zellen, Gewebe von Pflanzen oder Tieren, frisches oder gefrorenes biologisches Material enthalten, das die Chromatinstruktur bewahren kann, sind geeignet; Pore-C wurde erfolgreich für menschliche Zelllinien, Pflanzengewebe und Tiergewebe verwendet, wobei häufig eine geeignete Fixierung und Reinigung erforderlich ist, um die Interaktionssignale zu erhalten.
Q: Was sind typische Datenoutputs und Dateiformate aus der Pore-C-Analyse?
Der Pore-C-Workflow produziert rohe Long-Read-FASTQ- oder BAM/Concatemer-Reads, verarbeitete Kontaktpaare, Mehrweg-Interaktionsmatrizen (cooler/.mcool oder HiC-Stil), Methylierungsannotation und QC-Metriken wie gültige Kontakte pro Read, Fragmentanzahl sowie inter- versus intrachromosomale Kontaktverhältnisse.
Q: Wie viel Sequenzierungstiefe ist erforderlich, um mit Pore-C eine nützliche Genomassemblierung oder Interaktionskartierung zu erreichen?
Die erforderliche Tiefe hängt von der Genomgröße und -komplexität ab: Für die Scaffolding auf Chromosomenebene reicht oft eine moderate Abdeckung in Kombination mit Pore-C aus; für Telomer-zu-Telomer-Assemblierungen oder polyploide Genome verbessert eine höhere Abdeckung mit Langsequenzen und Pore-C die Ergebnisse; Pore-C erreicht im Allgemeinen ein Scaffolding, das mit einer viel höheren Tiefe von Hi-C vergleichbar ist, bei weniger Basen, wenn es richtig verarbeitet wird.
Q: Welche Bioinformatik-Workflows werden für die Pore-C-Datenanalyse verwendet?
Typische Pipelines verwenden einen "zuerst ausrichten, dann fragmentieren"-Ansatz, bei dem vollständige lange Reads auf das Referenzgenom abgebildet werden, dann Ligationsfragmente basierend auf den Schnittstellen von Restriktionsenzymen analysiert werden, multikontaktliche Informationen und paarweise Kontakte extrahiert werden, Kontaktkarten generiert, Methylierungsprofile erstellt, Qualitätskontrollen durchgeführt und Visualisierungen erstellt werden; Werkzeuge wie wf-pore-c werden häufig verwendet.
Pore-C Seq Fallstudien
1. Hintergrund
Traditionelles Hi-C erfasst paarweise Chromatin-Interaktionen, kann jedoch nicht auflösen Mehrwegkontakte oder Einzelallel-TopologieDiese Einschränkung behindert unser Verständnis von höherordentlichen 3D-Genomstrukturen und deren zelltyp-spezifische VariationPore-C und seine optimierte Hochdurchsatzversion (HiPore-C) ermöglichen die direkte Sequenzierung von Ketten, die mehrere ligierte DNA-Fragmente enthalten, und ermöglichen so die Entdeckung von Multi-Fragment-Interaktionen und epigenetische Informationen innerhalb einzelner langer Reads.
2. Methoden
Die Autoren wendeten HiPore-C auf Menschen an. GM12878 (B-Lymphozyt) und K562 (Erythroleukämie) Zellen. Verbesserungen in der Bibliotheksvorbereitung (Proteinase K + Pronase-Digestion) überwanden die Verstopfung der Nanoporen und erhöhten den Sequenzierungsausstoß. Anschließend integrierten sie Nanoporen-Sequenzierung mit dem MapPore-C-Pipeline, die Multi-Fragment-Reads auf das Referenzgenom abbildet und beide extrahiert Mehrweg-Interaktionen und DNA-Methylierungsmuster.
- Schlüsselmethode: HiPore-C mit ONT PromethION-Sequenzierung.
- Datenanalyse: Vergleich mit Hi-C, Clustering von Einzelallel-Topologien und gleichzeitige Methylierungsprofilierung.
3. Ergebnisse
- HiPore-C hat erfolgreich kanonische 3D-Genomstrukturen wie A/B-Kompartimente, TADs und Schleifen erfasst, mit hoher Übereinstimmung zu Hi-C (r = 0,958 für Eigenvektoren; r = 0,868 für Isolationswerte).
- Höherordentliche Interaktionen: ~38 % der Reads enthielten interchromosomale Fragmente, die aktive und inaktive Chromatin-Hubs aufdeckten (siehe Abbildung 3 auf Seite 5).
- Einzelallel-Auflösung: Reads, die in zelltyp-spezifische Topologien gruppiert sind, selbst in konservierten TADs (Abbildung 5 auf Seite 10).
- Funktionale Relevanz: Am β-Globin-Locus zeigte HiPore-C, dass Enhancer-Hubs nur in K562-Zellen gebildet werden, wo embryonale und fetale Globin-Gene exprimiert werden. In GM12878 blieb derselbe Locus stumm (Abbildung 6 auf Seite 12).
- Epigenetik: HiPore-C erfasste CpG-Methylierung genau, was mit WGBS übereinstimmte (r = 0,80), und verband sie mit Chromatinstrukturen (Abbildung 7 auf Seite 14).
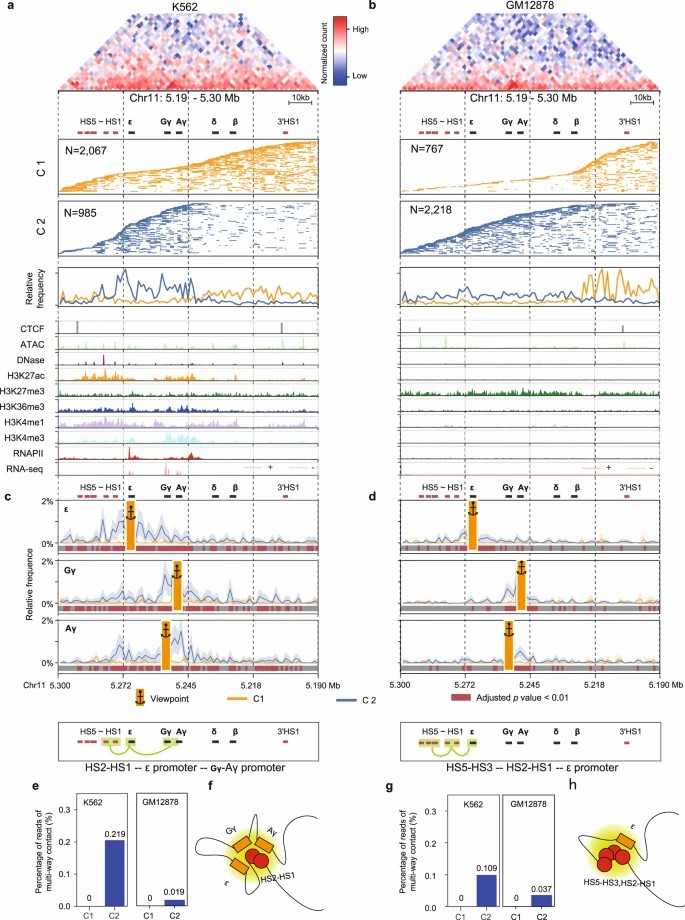 HiPore-C identifiziert ein zellspezifisches Enhancer-Hub am β-Globin-Locus und zeigt gleichzeitige Interaktionen zwischen Enhancern und fetalen Globin-Genen in K562-Zellen, jedoch nicht in GM12878-Zellen.
HiPore-C identifiziert ein zellspezifisches Enhancer-Hub am β-Globin-Locus und zeigt gleichzeitige Interaktionen zwischen Enhancern und fetalen Globin-Genen in K562-Zellen, jedoch nicht in GM12878-Zellen.
4. Schlussfolgerungen
HiPore-C bietet eine End-to-End-Lösung zur Erforschung der Genomfaltung auf Einzelallelauflösung, während gleichzeitig die DNA-Methylierung profiliert wird. Es offenbarte vielfältige Topologiecluster innerhalb von TADs, zelltypspezifische Enhancer-Promotor-Hubs und epigenetische Regulationen, die in der 3D-Genomarchitektur eingebettet sind. Dies zeigt sein Potenzial für funktionelle Genomik, Krankheitsforschung und die Assemblierung polyploider Genome.
Referenzen:
- Sean P. McGinty, Gulhan Kaya, View, Sheina B. Sim et al. CiFi: Präzise Langlese-Chromatin-Konformationsfängung mit geringen Eingabebedürfnissen.
- Zhang, Z., Yang, T., Liu, Y. u.a. Haplotyp-resolviertes Genom-Assembly und Resequenzierung bieten Einblicke in den Ursprung und die Züchtung moderner Rosen.. Nat. Pflanzes 10, 1659–1671 (2024).
- Tianyu Yang, Yifan Cai, Tianping Huang et al. Eine telomer-zu-telomer-gapfreie Referenzgenomassemblierung von Avocado bietet nützliche Ressourcen zur Identifizierung von Genen, die mit der Fettsäuresynthese und Krankheitsresistenz in Verbindung stehen., Gartenbau-Forschung, Band 11, Ausgabe 7, Juli 2024, uhae119,
- Jeon, D., Sung, YJ. & Kim, C. Hochwertige chromosomale Genomassemblierung des Wasabi (Eutrema japonicum) 'Magic'. Wissenschaftliche Daten 11, 1044 (2024).
- Jonghwan Choi, Taemin Kang, Sun-Jae Park, Seunggwan Shin, Eine chromosomengroße und annotierte Referenzgenomassemblierung von Plecia longiforceps Duda, 1934 (Diptera: Bibionidae), Genom Biologie und Evolution, Band 16, Ausgabe 10, Oktober 2024, evae205,
- Zhong, JY., Niu, L., Lin, ZB. et al. Hochdurchsatz-Pore-C zeigt die Einzelallel-Topologie und Zelltyp-Spezifität der 3D-Genomfaltung.. Nat Commun 14, 1250 (2023).


