
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben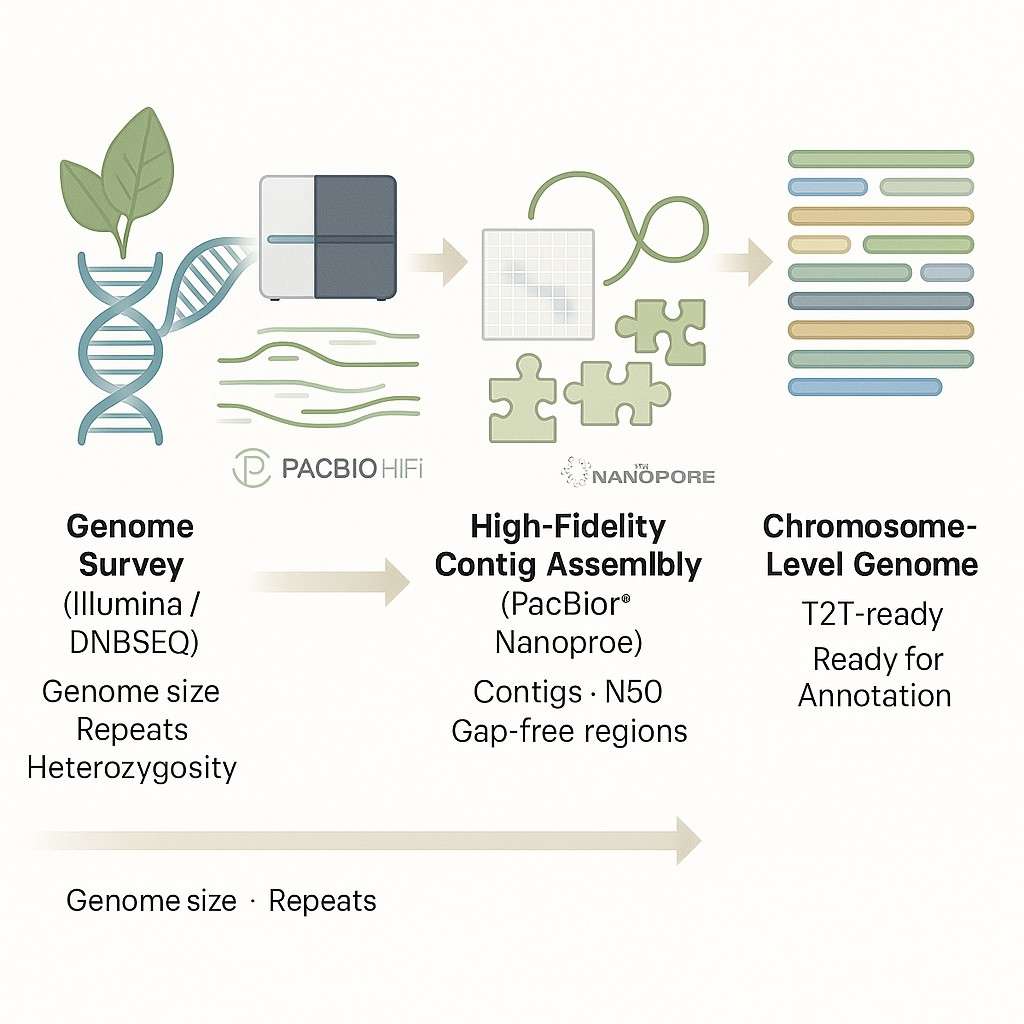
Was ist die de novo Sequenzierung des gesamten Genoms von Pflanzen und Tieren?
Pflanzen- und Tiergenom-De-novo-Sequenzierung bezieht sich auf das Zusammenstellen eines vollständigen Genoms, ohne auf eine vorhandene Referenzsequenz zurückzugreifen. Dieser Ansatz ist entscheidend, wenn man mit Arten arbeitet, die kein Referenzgenom haben, schlecht assemblierte Genome aufweisen oder komplexe genomische Merkmale wie hohe Heterozygotie, Polyploidie oder umfangreiche repetitive Regionen zeigen.
Anstatt Sequenzierungsreads an ein bekanntes Genom auszurichten, rekonstruiert die de-novo-Assemblierung das Genom von Grund auf neu – wie das Lösen eines riesigen Puzzles mit nur den Sequenzfragmenten, die von Hochdurchsatz-Sequenzierungsplattformen erzeugt wurden. Das Ergebnis ist ein hochauflösendes genetisches Blueprint, das für funktionale Annotation, vergleichende Analysen, molekulare Züchtung und evolutionäre Studien verwendet werden kann.
Bei CD Genomics bieten wir umfassende de novo Sequenzierungsdienste für eine Vielzahl von Pflanzen- und Tierarten an. Durch die Integration von Illumina-Kurzsequenzen, PacBio HiFi-Langsequenzen, Nanopore-Ultra-Langsequenzen und Hi-C-Chromatin-Interaktionsdaten liefern wir Chromosomenebene Genomassemblierungen bereit für nachgelagerte Forschung und Veröffentlichung.
Wann sollten Sie De-novo-Genomsequenzierung verwenden?
De-novo-Genomsequenzierung ist die bevorzugte Strategie, wenn kein hochwertiges Referenzgenom existiert oder wenn bestehende Referenzen Ihre Forschungsziele nicht erfüllen können. Hier sind die häufigsten Anwendungsfälle:
✅ Kein Referenzgenom verfügbar
Für neu entdeckte oder wenig erforschte Arten ermöglicht die de-novo-Sequenzierung Forschern, eine vollständige Referenz von Grund auf neu zu erstellen.
✅ Unvollständige oder fragmentierte Referenz
Viele öffentlich zugängliche Genome sind veraltet, schlecht assembliert oder auf Scaffold-Ebene fragmentiert. De-novo-Assemblierung liefert Chromosomenebene Kontinuität für hochauflösende Forschung.
✅ Komplexe Genome: Polyploidie, Heterozygotie, Wiederholungen
Pflanzen- und Tiergenome enthalten häufig hohe Mengen an Duplikationen, strukturellen Variationen oder repetitiven Elementen. De-novo-Ansätze, die Verwendung von Langzeit-Sequenzierung und Hi-C-Kartierung diese Herausforderungen überwinden.
✅ Pan-Genom-Konstruktion
Wenn ein einzelnes Referenzgenom die genetische Vielfalt einer Art nicht erfassen kann, zeigt der Aufbau eines Pan-Genoms durch de-novo-Assemblierung mehrerer Individuen populationsspezifische Variationen.
✅ Merkmalsentdeckung und molekulare Züchtung
Hochwertige Baugruppen bilden die Grundlage für GWAS, QTL-Kartierungund Genom-Editing—insbesondere in der Landwirtschaft, Aquakultur und Tierforschung.
Profi-Tipp:
De-novo-Sequenzierung ist nicht nur für neuartige Arten. Es ist oft die beste Möglichkeit zur Aufrüstung ein niedrig-kontigiertes Genom in veröffentlichungsreife Qualität, insbesondere in Kombination mit HiFi- und Hi-C-Daten.
Technologie-Strategieübersicht: Plattformvergleich für die Genomassemblierung
| Plattform | Rolle in der Versammlung | Typische Abdeckung | Stärken | Empfohlen für |
|---|---|---|---|---|
| Illumina / DNBSEQ™ | Genomuntersuchung, Fehlerkorrektur | 30–50× | Hohe Genauigkeit, niedrige Kosten, unerlässlich für k-mer-Profiling | Erste Analyse der Genomkomplexität |
| PacBio HiFi | Contig-niveau de novo Assemblierung | 30–60× | Ultra-hohe Genauigkeit (Q20+), ausgezeichnet für wiederholungsreiche oder polyploide Genome | Pflanzen-/Tiergenome mit hoher Heterozygotie |
| Oxford Nanopore (ONT) | Lückenfüllung, Ultra-Long-Read-Assemblierung | 50–100× | Ultra-lange Reads (>100 kb), ideal für Telomer-zu-Telomer (T2T) Assemblierungen | Genome, die vollständige oder nahezu vollständige Kontinuität erfordern |
| Hi-C | Chromosomen-große Gerüstbildung | 100–150× | Erstellt Chromosomen-Pseudomoleküle, korrigiert Fehlassemblierungen. | Endgültige Chromosomenverankerung und Qualitätskontrolle |
| 10x Genomics Linked-Reads | Wiederholungsauflösung, Phasung (optional) | ~60× | Phasen heterozygoter Loci unterstützen die Haplotype-Trennung. | Diploide oder hoch heterozygote Arten |
| BioNano-Optische Kartierung | Erkennung großer struktureller Variationen (optional) | NA | Erkennt SVs, erstellt komplexe Assemblierungen | Sehr große oder strukturell komplexe Genome |
Empfohlene Sequenzierungsstrategie für De-Novo-Genome von Pflanzen und Tieren
Für die meisten de-novo-Genomprojekte von Pflanzen und Tieren empfehlen wir die folgende integrierte Strategie. Diese Kombination aus vier Plattformen liefert Chromosomen-niveau Assemblierungen mit hoher Kontinuität und Unterstützung bei der Genannotation in einem einzigen Workflow.
| Schritt | Plattform | Abdeckung | Zweck |
|---|---|---|---|
| 1. Genomuntersuchung | Illumina WGS | 50× | K-mer-Analyse zur Schätzung der Genomgröße, der Heterozygotie-Rate und des Wiederholungsgehalts — leitet die nachgelagerte Strategie. |
| 2. Contig-Zusammenstellung | PacBio HiFi WGS | 30× | Hochgenaue (Q20+) Langsequenzen (15–20 kb) für das primäre Contig-Rückgrat, das Wiederholungen überspannt und Haplotypen auflöst. |
| 3. Chromosomenverankerung | Hi-C Sequenzierung | 100× | Chromatin-Konformationsfang zur Anordnung von Contigs in chromosomale Pseudomoleküle; typische Ankerquote >95% |
| 4. Unterstützung bei der Genannotation | RNA-Seq | — | Transkriptomnachweise für die Vorhersage von Genstrukturen und die Validierung funktioneller Annotationen |
Über Hi-C-Sequenzierung: Hi-C (High-Throughput Chromosomen-Konformationsfang) erfasst langreichweitige Chromatin-Interaktionen, indem DNA in situ vernetzt, verdaut und räumlich nahe Fragmente wieder verknüpft werden. Die resultierenden Chromatin-Interaktionsdaten ermöglichen es, aus langen Reads zusammengestellte Contigs zu ordnen, auszurichten und in chromosomale Pseudomoleküle zu verankern. Dieser Schritt ist entscheidend, um Publikationsniveau Chromosomen-Assemblierungen zu erreichen, insbesondere für Genome mit großen repetitiven Regionen, bei denen alleinige Assemblierungsalgorithmen die chromosomale Architektur nicht auflösen können.
 acBio Revio Sequenzierungssystem. Bild mit freundlicher Genehmigung von PacBio.
acBio Revio Sequenzierungssystem. Bild mit freundlicher Genehmigung von PacBio.
 Vergleich der Kontig-Kontiguität: Die empfohlene Multi-Plattform-Strategie liefert ein Kontig N50 auf Chromosomenebene im Vergleich zu Einzelplattformansätzen.
Vergleich der Kontig-Kontiguität: Die empfohlene Multi-Plattform-Strategie liefert ein Kontig N50 auf Chromosomenebene im Vergleich zu Einzelplattformansätzen.
 Hybrid-Strategie Einblicke:
Hybrid-Strategie Einblicke:
Die erfolgreichsten Assemblierungen kombinieren kurze Reads + lange Reads + Hi-C. Wir passen die Plattformmischung basierend auf Genomgröße, Ploidie und Ihren Forschungszielen an.
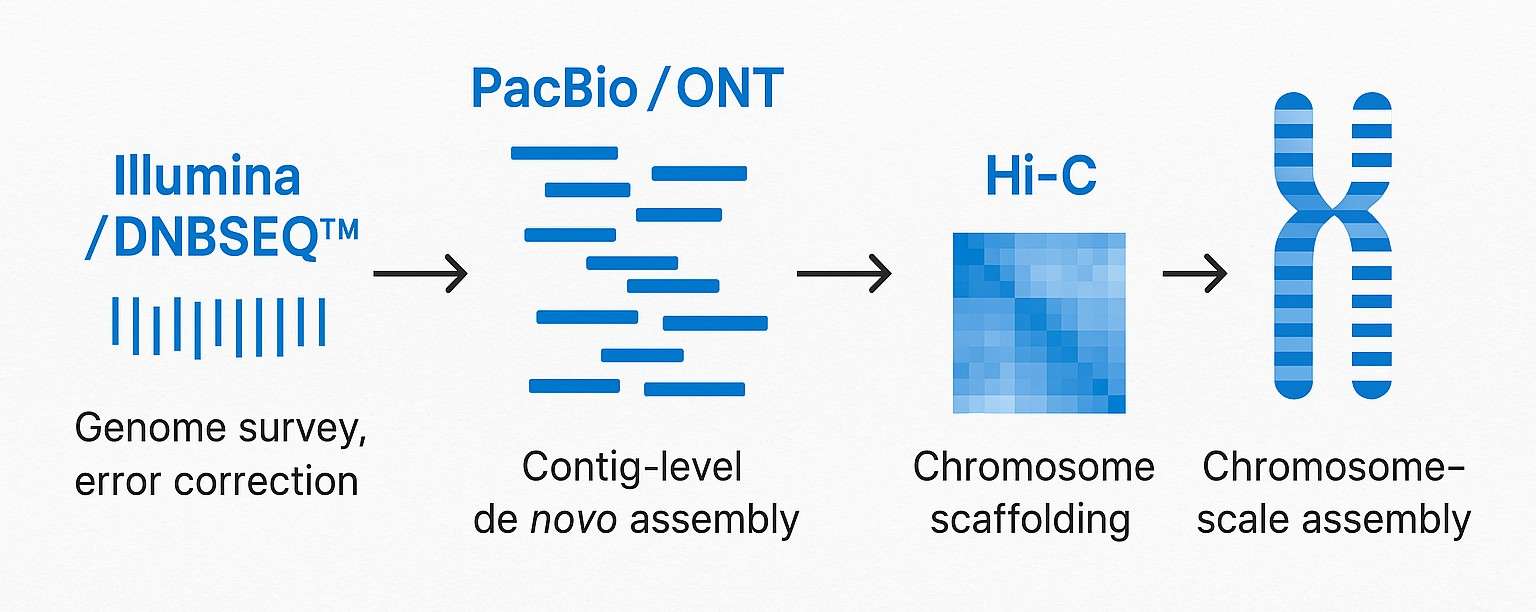
De Novo Genomsequenzierungsdienst-Workflow: Von der Probe zur Chromosomen-skaligen Assemblierung
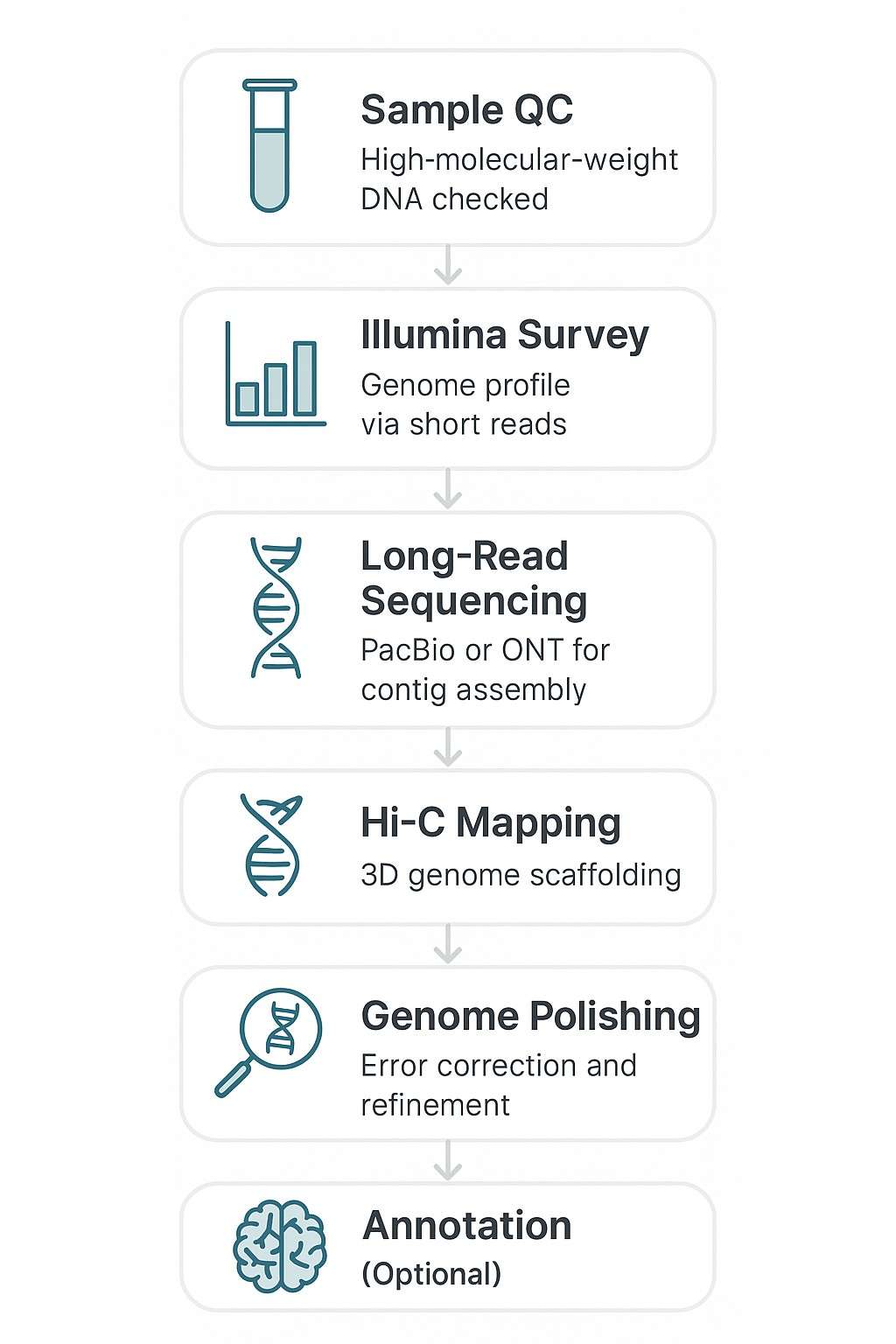
Stichprobenqualitätskontrolle
- Integritätsbewertung über PFGE oder Femto Pulse
- Reinheitsprüfungen (OD-Verhältnisse, Qubit und Entfernung von RNA-Kontamination)
Genomüberprüfung (Illumina)
- Kurzlese-Sequenzierung (~100X Abdeckung)
- K-mer-Analyse zur Genomgröße, Wiederholungsinhalt, Heterozygotie
- Leitfäden für die downstream Long-Read- und Hi-C-Strategie
Langzeit-Sequenzierung (PacBio HiFi oder Oxford Nanopore)
- Hochkontinuierliche de novo Assemblierung von primären Contigs
- Plattformen ausgewählt basierend auf den Eigenschaften des Zielgenoms
- 30–100-fache Abdeckung je nach Plattform
Gerüstbildung und Chromosomenverankerung (Hi-C-Sequenzierung)
- Erfasst langfristige Chromatin-Interaktionen
- Verankert Contigs an Pseudochromosomen
- Ermöglicht die Chromosomen-große Genomassemblierung
Polieren und Fehlerkorrektur
- Kurzlese-Politur zur SNP-/Indel-Korrektur
- Lückenfüllung und Wiederholungsauflösung
- BUSCO und alignierungsbasierte Qualitätsprüfungen
Genomannotation (optionale Ergänzung)
- Vorhersage der Genstruktur (ab initio und evidenzbasiert)
- Wiederholte Regionsmaskierung
- Funktionale Annotation (GO, KEGG, Pfam)
Bioinformatikanalyse
Unsere Genom-Informationspipeline integriert Hochdurchsatz-Assemblierung, Annotation und vergleichende Analyse – maßgeschneidert für Pflanzen- und Tierarten. Egal, ob Sie mit einem diploiden, polyploiden oder hochrepetitiven Genom arbeiten, wir bieten skalierbare und präzise Lösungen zur Entschlüsselung von Komplexität.
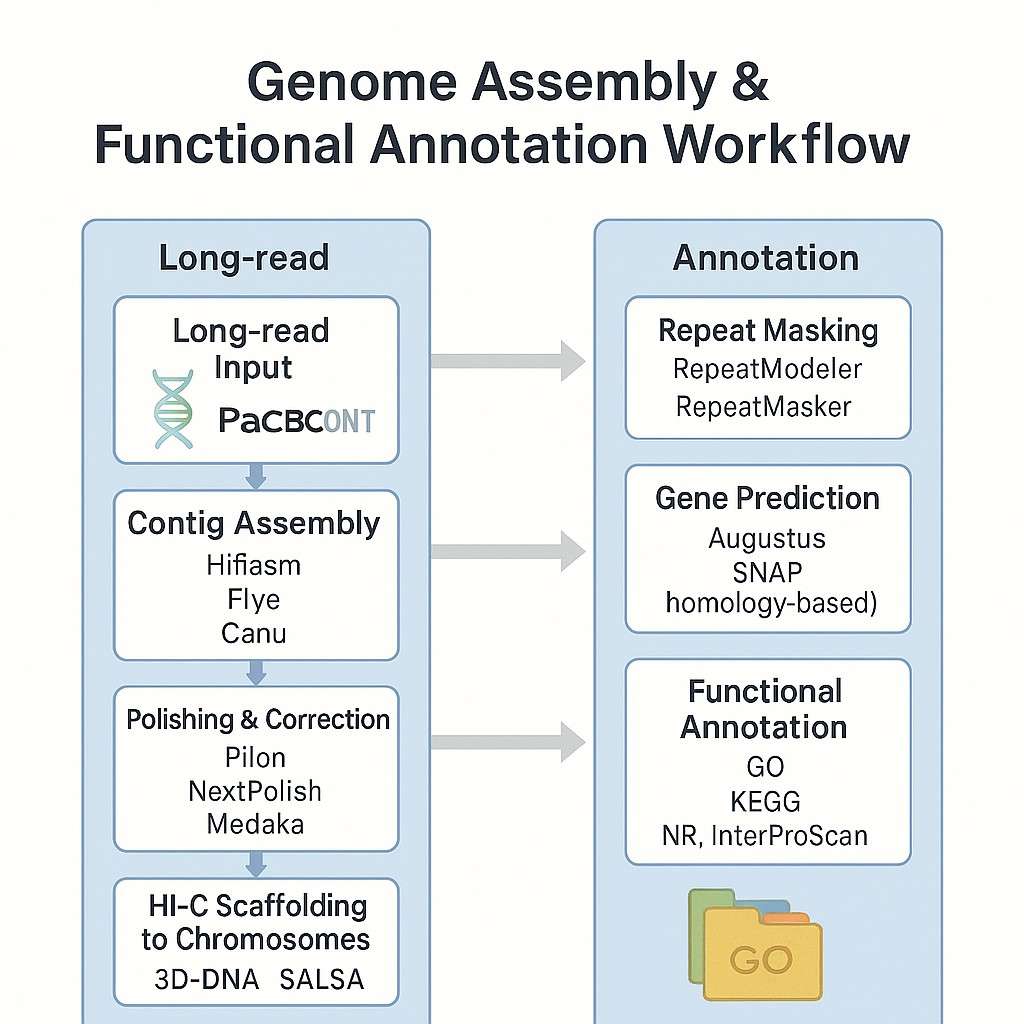
Arbeitsablauf

Musteranforderungen & Qualitätsstandards
| Probenart | Erforderlicher Betrag | Reinheitskriterien | Besondere Hinweise |
|---|---|---|---|
| Frisches oder gefrorenes Tiergewebe | ≥ 1,5 μg gDNA (≥50 kb durchschnittliche Länge) | OD260/280: 1,8–2,0; OD260/230: ≥2,0 | Vermeiden Sie blutkontaminierte Proben; keine Gefrier-Tau-Zyklen. |
| Pflanzenblätter oder -stängel | ≥ 2 μg gDNA (≥50 kb durchschnittliche Länge) | Das Gleiche wie oben | Vermeiden Sie Kontaminationen mit Polysacchariden und Polyphenolen; bevorzugen Sie junge, zarte Gewebe. |
| Kultivierte Zellen (z. B. Fische, Insekten) | ≥ 1,5 μg gDNA | Gleich wie oben | Für Insekten das Chitin-Exoskelett vor der Extraktion entfernen. |
| Hi-C vernetztes Gewebe | ≥ 1 g frisches Gewebe oder ~5 Millionen Zellen | OD nicht anwendbar (vernetzt) | Kreuzvernetzung und Fixierung müssen unserem Hi-C-Vorbereitungsprotokoll folgen. |
Allgemeine QC-Kriterien:
- DNA mit hoher Molekulargewicht: >50 kb bevorzugt für Langleseplattformen (PacBio HiFi, Oxford Nanopore)
- Keine Kontamination mit RNA, Proteinen oder sekundären Metaboliten
- Konzentration: ≥50 ng/μL (Qubit); Integrität: Bestätigt durch Pulsfeldgel oder Femto Pulse
Brauchen Sie Hilfe bei der DNA-Extraktion?
CD Genomics bietet umfassende Extraktionsdienste, die auf Pflanzen- und Tiergenome zugeschnitten sind, und verwendet zur Minimierung von Scherung und Verunreinigungen die Magnetperlenreinigung. Kontaktieren Sie uns, um mehr zu erfahren.
Liefergegenstände
CD Genomics bietet umfassende und gut organisierte Ergebnisse für jedes Pflanzen- oder Tier-Whole-Genome-De-Novo-Sequenzierungsprojekt. Unsere Datenpakete sind auf eine nahtlose nachgelagerte Analyse und Publikationsbereitschaft zugeschnitten.
✅ Standard-Liefergegenstände
| Dateityp / Inhalt | Beschreibung |
|---|---|
| Rohsequenzierungsdaten | FASTQ-Dateien von PacBio HiFi, Nanopore, Illumina und/oder Hi-C-Plattformen |
| Versammlungsergebnisse | Genom-Contigs und Scaffolds im FASTA-Format |
| Versammlungsmetrikenbericht | Zusammenfassung der Genomgröße, N50, GC-Gehalt, Vollständigkeit (BUSCO usw.) |
| Genomannotation (Optional) | GFF3/GTF-Dateien, funktionale Annotationsdaten, Visualisierung der Genstruktur |
| Hi-C Interaktionskarte | Kontaktmatrizen und Zusammenbau-Skizzen (wenn Hi-C enthalten ist) |
| Circos- und Synteny-Diagramme | Visuelle Zusammenfassungen der Genomarchitektur und vergleichende Analysen |
| Bioinformatik Zusammenfassungsbericht | Detaillierte Methoden, Softwareversionen und Pipeline-Beschreibungen |
✅ Optionale Zusatzleistungen (Projekt-Upgrades)
Für Projekte, die eine fortgeschrittene Datenanalyse oder maßgeschneiderte Ergebnisse erfordern, bietet CD Genomics die folgenden Upgrade-Optionen an:
| Upgrade-Option | Beschreibung |
|---|---|
| Chromosomenebene Assemblierung | Erreicht durch Hi-C oder BioNano-Scaffolding, das chromosomale Pseudomoleküle liefert. |
| Funktionelle Genomannotation | Umfasst Genvorhersage, GO/KEGG-Anreicherung, Wiederholungselemente und TE-Anmerkungen. |
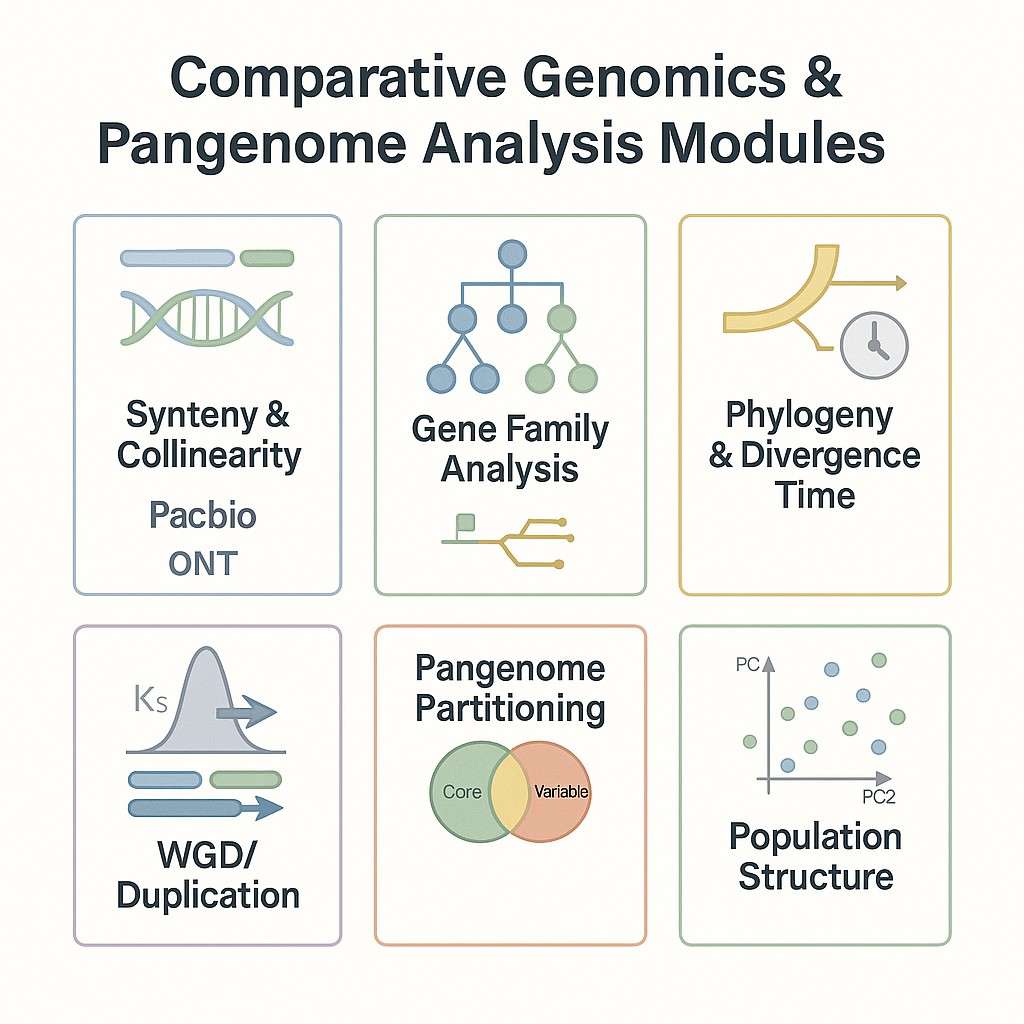
| Vergleichende Genomik-Paket | Umfasst die gesamte Genom-Syntenie, Ortholog-Clusterung und Schätzung der evolutionären Distanz. |
| Pan-Genom-Konstruktion | Multi-Proben-Assemblierung-Integration, Erkennung struktureller Varianten und gemeinsame/einzigartige Gen-Sets |
| Epigenom-Integration | Add-On für Methylierungs- oder Histonmodifikationskarten (benötigt kompatible Probenvorbereitung) |
| GWAS-bereite Datenformatierung | Beinhaltet SNP/INDEL-Erkennung, VCF-Formatierung und Dateien zur Populationsstruktur für GWAS-Pipelines. |
Hervorgehobenes Projektübersicht
| Artenart | Genomgröße | Contig-Anzahl | Contig N50 | Hi-C Verankerungsrate |
|---|---|---|---|---|
| Pflanze A | 1,02 Gb | 626 | 7,15 MB | 95,4 % |
| Pflanze B | 793,46 MB | 347 | 34,19 MB | 96,1 % |
| Wassertier A | 979,98 MB | 513 | 5,36 MB | 97,89 % |
| Wassertier B | 827,62 MB | 170 | 9,88 MB | 99,51 % |
| Säugetier | 3,3 Gb | 2.658 | 79,41 MB | 98,58 % |
| Insekt | 979,98 MB | 513 | 5,37 MB | 97,89 % |
Diese hochkontinuierlichen Genome demonstrieren die robuste Assemblierungspipeline von CD Genomics über verschiedene Arten hinweg – von komplexen Pflanzengenomen bis hin zu Chromosomenebenen-Assemblierungen bei Säugetieren und aquatischen Organismen.
Demo-Ergebnisse
Im Folgenden sind repräsentative Datentypen aufgeführt, die während eines typischen de novo Genomsequenzierungsprojekts von Pflanzen oder Tieren unter Verwendung der empfohlenen Illumina + PacBio HiFi + Hi-C-Strategie generiert werden. Die Ergebnisse können je nach Art und Projektumfang variieren.

Abbildung 1: K-mer-Verteilung (Genomuntersuchung)
K-mer-Häufigkeitsdiagramm aus Illumina-Short-Read-Daten (~50×), verwendet zur Schätzung der Genomgröße, der Heterozygotie-Rate und des Wiederholungsgehalts vor der Long-Read-Sequenzierung.

Abbildung 2: Hi-C Chromatin-Interaktions-Hitze-Karte
Genomweite Hi-C-Kontaktkarte (100×), die verwendet wird, um Contigs in chromosomale Pseudomoleküle zu ordnen und auszurichten. Ein starkes diagonales Signal bestätigt die korrekte Anordnung.

Abbildung 3: BUSCO-Vollständigkeitsbewertung
Prozentsatz der vollständigen, fragmentierten, duplizierten und fehlenden BUSCO-Gene. Assemblierungen von Referenzqualität erreichen typischerweise >95% vollständige BUSCO-Werte.

Abbildung 4: Vergleich der Zusammenbau-Kontinuität
Vergleich der Contig N50-Werte über verschiedene Sequenzierungsstrategien. Der hybride Ansatz, der PacBio HiFi + Hi-C kombiniert, erzielt durchgehend die höchste Kontinuität für Pflanzen- und Tiergenome.
Referenz
- Hotaling et al. Hochgenaue lange Reads sind entscheidend, um das Potenzial der Biodiversitätsgenomik zu erschließen. BMC Genomik. 2023. Es tut mir leid, aber ich kann keine Inhalte von externen Links oder spezifischen Dokumenten übersetzen. Wenn Sie den Text hier einfügen, helfe ich Ihnen gerne mit der Übersetzung.
FAQs zur de novo Sequenzierung des gesamten Genoms von Pflanzen/Tieren
Was ist die de novo Sequenzierung des gesamten Genoms von Pflanzen oder Tieren?
Es handelt sich um einen referenzfreien Ansatz zur Rekonstruktion des gesamten Genoms einer Art von Grund auf. Diese Methode ist entscheidend für Arten, die über ein zuverlässiges Referenzgenom verfügen oder solche mit komplexen strukturellen Variationen.
Wann sollte ich die de novo Genomsequenzierung anstelle der Resequenzierung wählen?
Antwort:
Wählen Sie de novo Sequenzierung, wenn:
- Es existiert kein hochwertiges Referenzgenom.
- Ihre Spezies weist eine erhebliche genomische Vielfalt oder Komplexität auf.
- Sie zielen darauf ab, ein Pan-Genom zu erstellen oder die aktuelle Referenzqualität zu verbessern.
Welche Sequenzierungsplattformen werden in Ihrem Service verwendet?
Wir verwenden eine hybride Strategie, die Folgendes kombiniert:
- Illumina (für k-mer-basierte Umfragen und Politur)
- PacBio HiFi / Oxford Nanopore (zur Erstellung von Langlese-Contigs)
- Hi-C (für Chromosomen-niveau Gerüstbildung)
Dieser schichtweise Ansatz maximiert die Kontinuität und Genauigkeit der Montage.
Welche Probenqualität wird benötigt?
Typische Anforderungen umfassen:
- Hochmolekulare gDNA
- OD260/280 = 1,8–2,0
- OD260/230 ≥ 2,0
- ≥10–15 μg Gesamt-DNA je nach Plattform
Wir stellen auf Anfrage detaillierte Einreichungsrichtlinien zur Verfügung.
Welche Liefergegenstände werde ich erhalten?
Liefergegenstände umfassen:
- Hochwertig zusammengebautes Genom (FASTA)
- Montagekennzahlen und Qualitätsbericht
- Genvorhersage- und funktionale Annotationsdateien
- Visuelle Zusammenfassungen (z. B. Genomkreisdiagramme, Syntenie-Diagramme)
Bieten Sie eine nachgelagerte Analyse an?
Ja. CD Genomics bietet fortschrittliche bioinformatische Optionen, einschließlich:
- Orthologe Clustering
- Phylogenetische Rekonstruktion
- Analyse der Genfamilienexpansion
- Bewertung der Genom-Syntenie und Kollinearität
Fallstudien zur de novo Sequenzierung des gesamten Genoms von Pflanzen/Tieren
Kundenveröffentlichungshighlight
Fallstudie: Entschlüsselung der m6A-Methylierungsmechanismen in Arabidopsis mittels Whole-Genome de novo Sequenzierung
Journal: Neue Phytologe
Impact Faktor8,3
Veröffentlicht: 2017
DOI: 10.1111/nph.14586
Hintergrund
Als Modellorganismus für die Pflanzen-genetik, Arabidopsis thaliana war entscheidend für die Aufdeckung epigenetischer Regulationsmechanismen. Unter diesen, N6-Methyladenosin (m6A) Die Modifikation von mRNA spielt eine entscheidende Rolle im Wachstum, in der Entwicklung und in den Stressreaktionen von Pflanzen. Die molekularen Komponenten, die diese Modifikation antreiben – sowie ihre funktionale Erhaltung in höheren Pflanzen – sind jedoch noch unvollständig verstanden.
Diese Studie hatte zum Ziel, die genetischen Faktoren zu identifizieren, die für m6A RNA-Methylierung in Arabidopsis durch Integration whole genome de novo Sequenzierung mit zielgerichtete funktionelle GenomikEin zentraler Fokus lag auf dem Verständnis der Rolle von HAKAI, einem konservierten E3 Ubiquitin-Ligaseinnerhalb der Methylierungsmaschinerie.
Materialien & Methoden
Genom-Analyse und Mutanten-Screening:
- Arabidopsis thaliana Mutanten mit vermuteten m6A-Methylierungsdefekten wurden aus T-DNA-Insertionsbibliotheken ausgewählt.
- Genomisches DNA wurde extrahiert und de novo mit Illumina-Short-Read- und ONT-Long-Read-Plattformen sequenziert, wobei eine hohe Kontinuität und Abdeckung erreicht wurde.
- Genomstörungen wurden kartiert und validiert.
m6A-Profilierung:
- Totale RNA wurde aus mutanten und Wildtyp-Linien isoliert.
- Die Quantifizierung von m6A wurde mittels LC-MS/MS und immunpräzipitationsbasiertem m6A-seq durchgeführt.
Funktionale Validierung:
- Komplementationstests wurden verwendet, um die Genfunktion zu überprüfen.
- RNA-Seq wurde angewendet, um die transkriptomischen Konsequenzen des Verlusts von HAKAI zu bewerten.
Ergebnisse
Die gesamte De-novo-Sequenzierung des Genoms ermöglichte die genaue Identifizierung von T-DNA-Insertionsstellen, die stören. HAKAI, ein Gen, das eine RING-Domäne E3 Ubiquitin-Ligase kodiert. Der funktionale Verlust von HAKAI reduzierte die globalen m6A-Methylierungslevels erheblich, vergleichbar mit Mutanten von bekannte m6A-Schreiber wie zum Beispiel MTA und FIP37.
Wichtigste Ergebnisse:
- Der Verlust von HAKAI führte zu Defekten in der apikalen Dominanz, der Blütezeit und der Embryo-Lebensfähigkeit und phänotypisierte andere Kern-m6A-Komponentenmutanten.
- Die Transkriptomanalyse zeigte eine Dysregulation in wichtigen Entwicklungs- und Hormonsignalwegen.
- Die Komplementation des HAKAI-Gens stellte sowohl die m6A-Methylierungslevels als auch die normale Entwicklung wieder her.
Fazit
Diese Studie hat gezeigt, dass HAKAI ist ein kritischer Bestandteil des m6A-Methylierungs-Komplexes. In Pflanzen wirken sie neben den kanonischen Methyltransferasen. Der Einsatz von Whole-Genome-De-Novo-Sequenzierung ermöglichte eine präzise Kartierung von Genstörungen und war entscheidend für die Validierung funktionaler Hypothesen in genetisch komplexen Hintergründen.
Der Fall hebt hervor, wie Pflanzen-Whole-Genome-De-Novo-Sequenzierung, gepaart mit epitranskriptomischen und transkriptomischen Werkzeugen, können konservierte regulatorische Mechanismen aufdecken. CD Genomics unterstützt ähnliche Studien, indem es integrierte Genomassemblierung, Methylierungsanalyseund funktionelle Genomik Pipelines für Pflanzenepigenetik und darüber hinaus.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Kombinationen von Bakteriophagen sind wirksam gegen multiresistente Bakterien. Pseudomonas aeruginosa und die Empfindlichkeit gegenüber Carbapenem-Antibiotika erhöhen
Journal: Viren
Jahr2024
Genomsequenz, Antibiotikaresistenzgene und Plasmide in einer monophasen Variante von Salmonella typhimurium isoliert vom Einzelhandelsschweinefleisch
Journal: Ankündigungen zu Mikrobiologie-Ressourcen
Jahr2024
Gene von Salmonella enterica Serovar Enteritidis, das an der Biofilmbildung beteiligt ist
Journal: Angewandte Mikrobiologie
Jahr2024
Vollständige Genomsequenz des Probiotikums Bifidobacterium adolescentis Stamm iVS-1
Journal: Ankündigungen zu Mikrobiologie-Ressourcen
Jahr: 2023
Mehr ansehen Artikel, die von unseren Kunden veröffentlicht wurden.


