
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
RIP-Seq
CD Genomics bietet fortschrittliche RIP-seq-Dienste an, die nutzen Next-Generation-Sequenzierung (NGS) Technologie. Unser RIP-seq-Service bietet eine präzise und umfassende Analyse von RNA-Protein-Interaktionen und liefert detaillierte Einblicke in RNA-bindende Proteininteraktionen über verschiedene biologische Proben hinweg.
Die Einführung in RIP-Seq
Jede Aktivität des Transkriptlifecycles, von der Entstehung (Polymerasen) bis zum Abbau (Nukleasen), umfasst die Bindung von Proteinen. Neben der Proteinproduktion spielen eine Teilmenge dieser Transkripte entscheidende Rollen in anderen wesentlichen Prozessen wie der epigenetischen Regulation und dem Genomschutz durch Transposonsilencing. Die meisten Studien haben sich auf die Transkriptom-Profilierung konzentriert. Es wird jedoch angenommen, dass die Mengen an mRNAs nicht immer direkt mit den stabilen Proteinspiegeln korrelieren. Das Interesse an der Identifizierung der RNAs, die mit RNA-bindenden Proteinen (RBP) in einem zellulären Kontext assoziiert sind, wächst, da die Rolle der RNA-Verarbeitung und der translationalen Ereignisse, die posttranskriptionell stattfinden, zunehmend anerkannt wird.
RIP-Seq kartiert die Stellen, an denen Proteine an die RNA innerhalb von RNA-Protein-Komplexen gebunden sind. Die RNA-Immunpräzipitation (RIP) impliziert die Reinigung von RNA-Protein-Interaktionen unter nativen Bedingungen durch die Verwendung eines protein-spezifischen Antikörpers, um das interessierende RBP zu kartieren. Das Aufkommen von Sequenzierungstechnologien, kombiniert mit verschiedenen RIP-Chemien, hat die gleichzeitige Erkennung von Tausenden gebundener Transkripte (mRNAs, nicht-kodierende RNAs oder virale RNAs) in einem einzigen Experiment ermöglicht. CD-Genomik bietet RIPed RNA-Sequenzierung, um Einblicke nicht nur in die gut etablierten Prozesse wie Transkription, Spleißen und Translation zu erhalten, sondern auch in neuere Bereiche wie RNA-Interferenz und Genregulation durch nicht-kodierende RNAs.
Vorteile unseres RIP-Seq-Services
- In der aktuellen biologischen Forschung hebt sich RIP-seq als die effektivste Methode zur Bestimmung von Protein-RNA-Interaktionen im natürlichen Zustand einer Zelle hervor. Diese Technik identifiziert effizient, ob ein Protein ein RNA-bindendes Protein ist, erläutert, welche RNAs direkt mit dem Protein interagieren, und lokalisiert deren Bindungsstellen.
- RIP-seq ermöglicht die umfassende Untersuchung von Protein-RNA-Interaktionen auf der Ebene des gesamten Transkriptoms und gibt Aufschluss über die Arten von RNAs, die an diesen Interaktionen beteiligt sind.
- Mit seiner hohen Auflösung bietet RIP-seq die Möglichkeit, RNA-Sequenzen zu erkennen, die mit Proteinen interagieren, und liefert letztendlich wertvolle Einblicke in die komplexe Landschaft der Protein-RNA-Interaktionen.
- Kosten-Effektivität: Der experimentelle Zyklus ist kurz, die Analyse umfassend und der Preis niedrig.
- Hohe Abdeckung: Das gesamte Transkriptom ist abgedeckt, was das Screening und die Identifizierung von Proteinbindungsstellen im gesamten Transkriptom ermöglicht.
- Hohe Empfindlichkeit: Aus jeder Probe können Millionen von Sequenz-Tags gewonnen werden, was die Entdeckung seltener Proteinbindungsstellen im Transkriptom ermöglicht.
- Hohe Präzision: Daten mit hohem Signal-Rausch-Verhältnis können gewonnen werden, die wahre Ereignisse präzise von Rauschen unterscheiden und Proteinbindungsstellen genau lokalisieren.
- Angepasster RNA-Immunpräzipitation-Sequenzierungsplan: Maßgeschneiderte RNA-Immunpräzipitation-Sequenzierungspläne können basierend auf spezifischen Bedürfnissen erstellt werden.
Anwendungen von RIP-Seq
- Untersuchung der Wechselwirkungen zwischen RNA und Proteinen in Zellen
- Identifizierung der Wechselwirkungen zwischen RBPs und nicht-kodierenden RNAs (wie LncRNAs, miRNAs usw.)
- Kartierung der genomweiten Interaktionen zwischen RNA und RBPs
RIP-Seq-Workflow
CD Genomics wendet die Illumina-Technologie an. NGS Ausrüstung zur Sequenzierung der gebundenen Transkripte, die genomweite Interaktionen von RBPs aufdeckt. Forscher werden die RNA einreichen, die durch die entsprechenden RIP-Methoden basierend auf spezifischer Proteinimmunpräzipitation aus Zelllinien oder Geweben gewonnen wurde. Unser RIP-Seq-Service, der den Workflow von der Proben-QC bis zur Datenanalyse anbietet, ermöglicht eine schnelle Profilierung und tiefgehende Einblicke in die RNA.
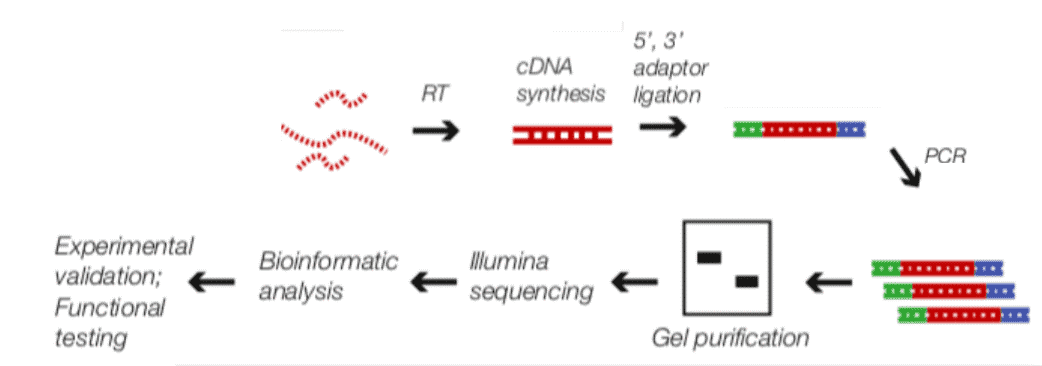 Abb. 2. RIP-Seq Arbeitsablauf
Abb. 2. RIP-Seq Arbeitsablauf
Dienstspezifikationen
Musteranforderungen
|
|
 Klicken |
Sequenzierungsstrategie
|
 |
Bioinformatikanalyse Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an:
|
Analyse-Pipeline
Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details in RIP-Seq für Ihr Schreiben (Anpassung)
Wenn Sie zusätzliche Anforderungen oder Fragen haben, zögern Sie bitte nicht, uns zu kontaktieren.
Referenz
- Zhao J u. a.Genomweite Identifizierung von polycomb-assoziierten RNAs durch RIP-seq. Molekulare Zelle, 22. Dez. 2010;40(6):939-53
Demo-Ergebnisse
Teilweise Ergebnisse sind unten aufgeführt:
RIP-Seq häufig gestellte Fragen
1. Was sind einige aktuelle Fortschritte in der RIP-seq?
Die jüngsten Fortschritte in der RIP-seq-Technologie haben sich erheblich weiterentwickelt. Diese Entwicklungen umfassen Verbesserungen in der Antikörpertechnik mit Fokus auf Spezifität, verfeinerte Kreuzvernetzungstechniken, die eine überlegene Isolation von RNA-Protein-Komplexen unterstützen, sowie die Einführung ausgeklügelter bioinformatischer Ressourcen zur Dateninterpretation. Darüber hinaus bietet die Integration von RIP-seq mit Methoden wie CLIP-seq (Kreuzvernetzung und Immunpräzipitation-Sequenzierung) zusätzliche Perspektiven auf RNA-Protein-Interaktionen.
2. Wie kann der Erfolg von RIP-seq-Experimenten sichergestellt werden?
Antikörpervalidierung: Verwenden Sie gut charakterisierte, hochaffine Antikörper.
Optimierte Methoden: Halten Sie sich an optimierte Verfahren für Zelllyse, Immunpräzipitation und RNA-Extraktion.
Geeignete Kontrollen: Integrieren Sie geeignete Kontrollen (wie unspezifische IgG-Kontrollen, Eingangs-RNA), um spezifische Interaktionen von Hintergrundsignalen zu unterscheiden.
Replikation: Führen Sie biologische Replikate durch, um die Reproduzierbarkeit der Ergebnisse zu bestätigen.
Datenintegrität: Verwenden Sie hochwertige Sequenzierungsplattformen und gründliche Bioinformatik Analysen zur Gewährleistung der Datengenauigkeit und -zuverlässigkeit.
3. Was ist Input und welche Rolle spielt er in Experimenten?
Nach der RNA-Fragmentebildung muss vor der Immunpräzipitation ein Teil der Probe als Input-Kontrolle beiseitegelegt werden (nicht der Immunpräzipitation unterzogen). Der Input besteht aus der fragmentierten RNA, die zusammen mit der immunpräzipitierten RNA der Probe einer Rückkreuzvernetzung, RNA-Reinigung und anschließenden PCR oder anderen Nachweismethoden unterzogen wird. Durch die anschließende Datenanalyse ermöglicht die Input-Kontrolle den Ausschluss von Hintergrundgeräuschen (falsch positiven Peaks, die durch unspezifische Bindung verursacht werden), validiert die Effizienz der RNA-Fragmentierung und bestätigt die IP-Effizienz während des gesamten Experiments. Daher ist die Input-Kontrolle ein unverzichtbarer Schritt in IP-seq-Experimenten.
RIP-Seq Fallstudien
RIP-Seq von EZH2 identifiziert TCONS-00036665 als Regulator der Myogenese bei Schweinen
Zeitschrift: Frontiers in Cell and Developmental Biology
Impact-Faktor: 6,081
Veröffentlicht: 12. Januar 2021
Hintergrund
Die Myogenese der Wirbeltier-Skelettmuskulatur umfasst Vorläuferzellen, die von Pax3 und Pax7 reguliert werden, welche Myf5 und MyoD zur Muskel-Differenzierung induzieren. Satellitenzellen unterstützen die Regeneration. EZH2, ein Polycomb-Protein, ist entscheidend für die Muskelentwicklung und die Genregulation durch Histon-Methylierung. lncRNAs interagiert mit EZH2, um die Myogenese zu regulieren. Diese Studie verwendete RIP-Seq in Kombination mit lincRNA-Sequenzierung (lincRNAseq), um 356 neuartige lincRNAs im Skelettmuskel von Schweinen zu identifizieren, und enthüllte Einblicke in ihre Rollen, wie TCONS-00036665, das die Proliferation von Satellitenzellen fördert, aber die Differenzierung hemmt, wodurch die Muskelentwicklung beeinflusst wird.
Materialien & Methoden
Probenvorbereitung
- Reine Large White weibliche Schweine
- Muskelgewebe des Longissimus dorsi
- C57-Mäuse
- Gesamte RNA-Extraktion
Sequenzierung
- RIP-Seq
- LincRNAseq
- ChIP-Seq
- Illumina HiSeq 2000
- Qualitätskontrolle
- Ausrichtung
- Transkriptzusammenstellung
- Ausdrucksanalyse
- Statistische Analyse
Ergebnisse
In dieser Studie identifizierte RIP-Seq EZH2-bindende lincRNAs aus dem Skelettmuskel von 1-monatigen Schweinen. Mithilfe einer durch Western Blot validierten EZH2-Antikörperanreicherung führten die Autoren RIP-Seq und lincRNAseq durch. Filterungsschritte basierend auf Expressionsniveaus und CPC-Werten identifizierten 356 EZH2-bindende lincRNAs, die überwiegend in intergenen Regionen lokalisiert sind, was auf ihre regulatorischen Rollen in der Muskelbiologie hindeutet.
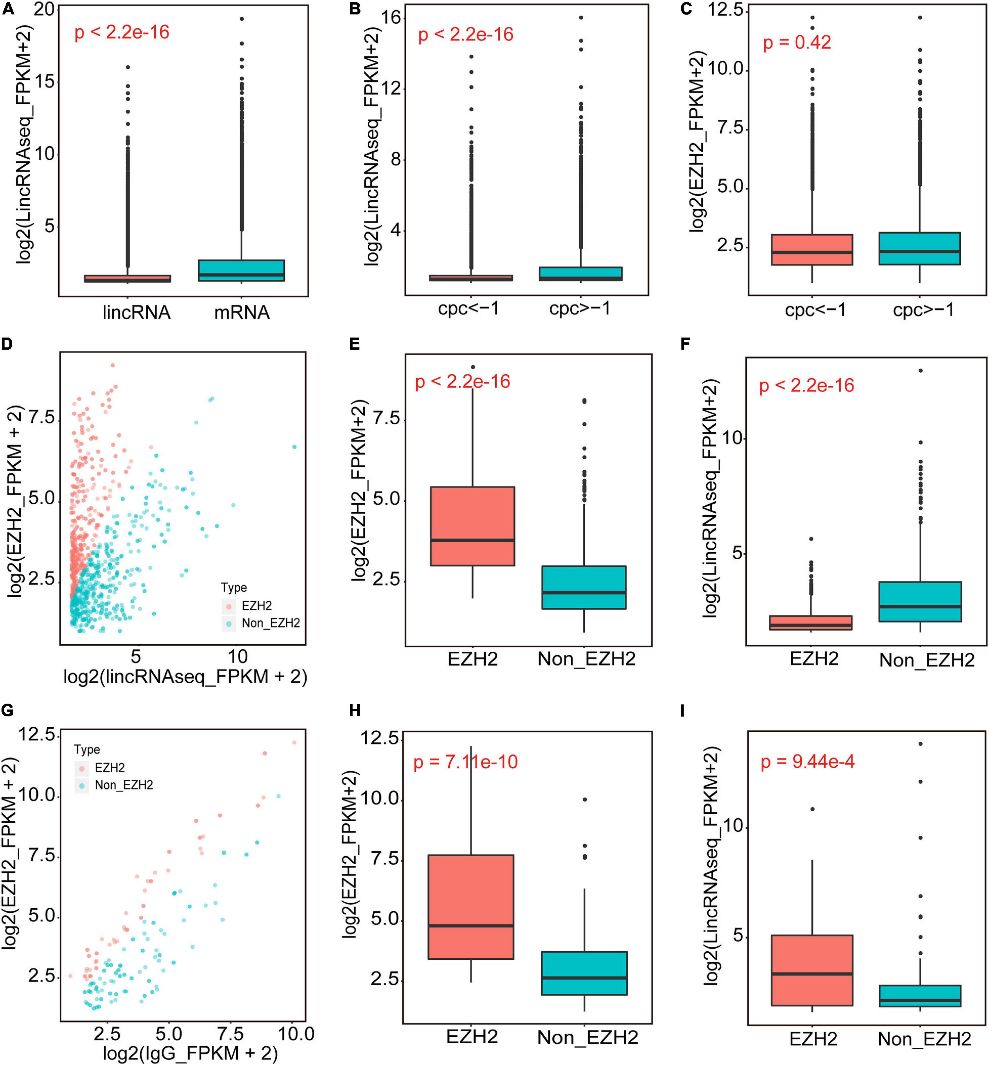 Abbildung 1. Die Identifizierung neuartiger lincRNAs, die an EZH2 binden.
Abbildung 1. Die Identifizierung neuartiger lincRNAs, die an EZH2 binden.
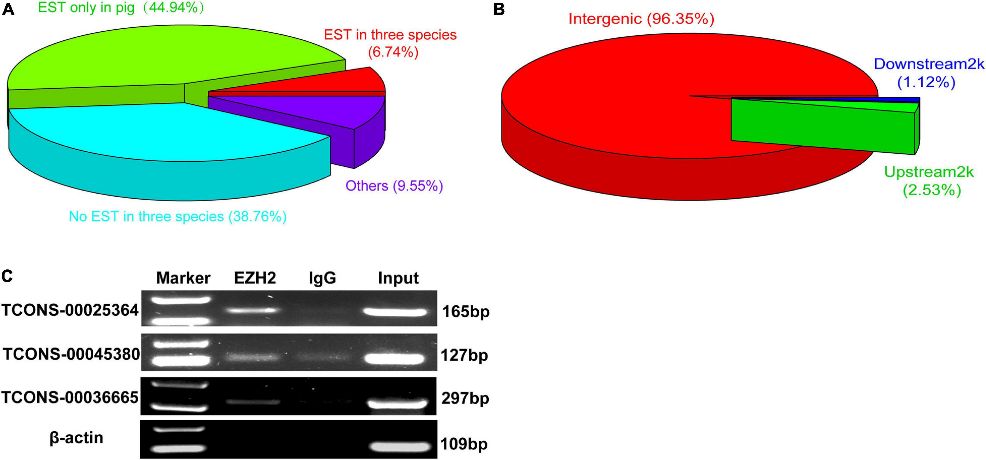 Abbildung 2. Die Verifizierung neuartiger lincRNAs, die an EZH2 binden.
Abbildung 2. Die Verifizierung neuartiger lincRNAs, die an EZH2 binden.
Drei lincRNAs, TCONS-00025364, TCONS-00045380 und TCONS-00036665, wurden durch RIP-PCR-Experimente bestätigt, dass sie an EZH2 binden (Abbildung 2C). TCONS-00036665, das mit dem Schwein EF397601-Transkript übereinstimmt, weist Ähnlichkeiten mit dem menschlichen und maus NEAT1-Gen auf, was auf seine Beteiligung an der Entwicklung der Skelettmuskulatur hindeutet. Weitere Charakterisierungen zeigten, dass TCONS-00036665 ein 3.450 bp polyadenyliertes Transkript ist, das überwiegend in den Zellkernen von proliferierenden und differenzierten Muskel-Satellitenzellen (PSCs) des Schweins lokalisiert ist. Die Expressionsanalyse ergab, dass TCONS-00036665 während der Differenzierung der PSCs zunimmt, was auf seine Rolle bei der Regulierung von Proliferations- und Differenzierungsprozessen hindeutet.
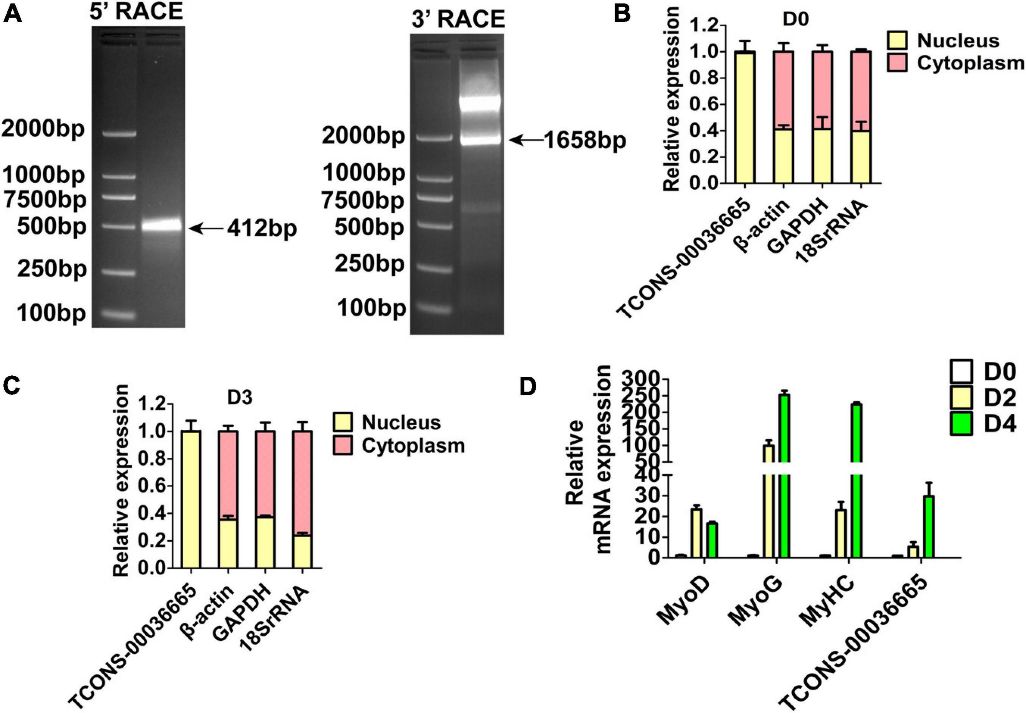 Abbildung 3. Die molekulare Charakterisierung von TCONS-00036665.
Abbildung 3. Die molekulare Charakterisierung von TCONS-00036665.
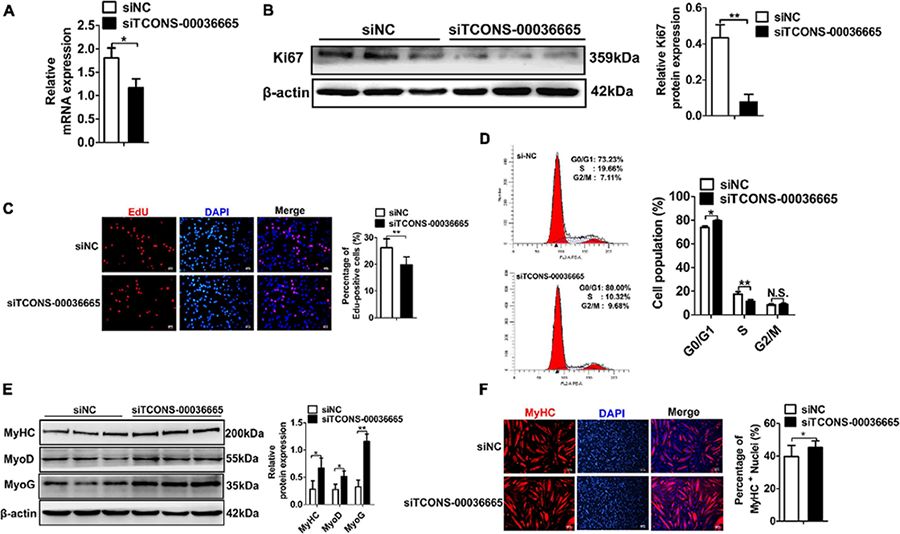 Abbildung 4. Der Knockdown von TCONS-00036665 hemmt die Proliferation von PSC, fördert jedoch die Differenzierung von PSC.
Abbildung 4. Der Knockdown von TCONS-00036665 hemmt die Proliferation von PSC, fördert jedoch die Differenzierung von PSC.
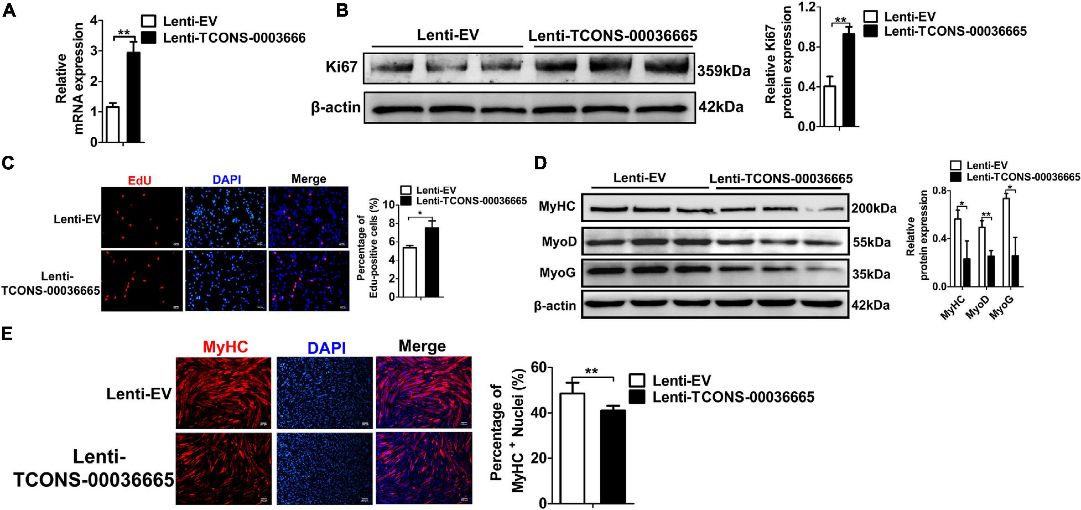 Abbildung 5. Die Überexpression von TCONS-00036665 fördert die Proliferation von PSC, hemmt jedoch die Differenzierung von PSC.
Abbildung 5. Die Überexpression von TCONS-00036665 fördert die Proliferation von PSC, hemmt jedoch die Differenzierung von PSC.
Fazit
In dieser Studie wurde die von EZH2 vermittelte Genrepression im Skelettmuskel von Schweinen untersucht. TCONS-00036665, eine EZH2-bindende lincRNA, wurde identifiziert und stellte sich als Regulator der Proliferation und Differenzierung von Muskel-Satellitenzellen heraus, indem sie die EZH2-vermittelte Anreicherung von H3K27me3 an den Promotoren von Zielgenen verstärkt. Die Ergebnisse deuten auf eine neuartige regulatorische Rolle von lincRNAs in der Muskelentwicklung von Schweinen durch epigenetische Mechanismen, die EZH2 einbeziehen, hin.
Referenz
- Wang S, Xu X, Liu Y, et al. RIP-Seq von EZH2 identifiziert TCONS-00036665 als Regulator der Myogenese bei Schweinen. Frontiers in Zell- und Entwicklungsbiologie, 2021, 8: 618617.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Die Spaltung von Phagen-DNA durch Restriktionsendonukleasen ermöglicht die Wiederbelebung aus der durch Cas13 induzierten bakteriellen Dormanz.
Zeitschrift: Nature Mikrobiologie
Jahr: 2023
IL-4 fördert die Erschöpfung von CD8.+ CART-Zellen
Zeitschrift: Nature Communications
Jahr: 2024
Fettreiche Diäten während der Schwangerschaft führen zu Veränderungen der DNA-Methylierung und Proteinexpression im Pankreasgewebe der Nachkommen: Ein Multi-Omics-Ansatz
Internationales Journal für Molekulare Wissenschaften
Jahr: 2024
KMT2A assoziiert mit dem PHF5A-PHF14-HMG20A-RAI1 Subkomplex in Stammzellen des Bauchspeicheldrüsenkrebses und reguliert epigenetisch deren Eigenschaften.
Zeitschrift: Naturkommunikationen
Jahr: 2023
Krebsassoziierte DNA-Hypermethylierung von Polycomb-Zielen erfordert die doppelte Erkennung von Histon H2AK119-Ubiquitinierung und der sauren Tasche des Nukleosoms durch DNMT3A.
Journal: Wissenschaftliche Fortschritte
Jahr: 2024
Genomisches Imprinting-ähnliches monoalleles väterliches Ausdrucksmuster bestimmt das Geschlecht von Kanalkatzenfischen.
Journal: Wissenschaftliche Fortschritte
Jahr: 2022
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


