
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Genexpressionsprofilierung Mikroarray-Service
CD Genomics bietet hochwertige Dienstleistungen für mikroarray-basierte Transkriptomstudien zu einem sehr erschwinglichen Preis an.
Was ist ein Mikroarray zur Genexpressionsprofilierung?
Genexpressions-Mikroarrays stellen ein fortschrittliches technologisches Wunder dar, das es Wissenschaftlern ermöglicht, die Expressionsniveaus von Tausenden von Genen gleichzeitig zu messen. Ursprünglich in den mittleren 1990er Jahren entwickelt, hat diese Innovation unser Verständnis der Genexpression dramatisch verändert, indem sie umfassende, genomweite Analysen ermöglicht.
Ein Mikromatrix ähnelt einer festen Leinwand, sei es ein Glasobjektträger oder ein Siliziumchip, der sorgfältig mit Tausenden von DNA-Sonden angeordnet ist. Diese DNA-Sonden sind so strukturiert, dass sie mit komplementären mRNA-Sequenzen hybridisieren, die aus einer Probe extrahiert wurden. Dadurch wird die präzise Erkennung und Quantifizierung von Genexpressionsniveaus ermöglicht.
Der Prozess der Nutzung Mikroarray Die Technologie zur Genexpressionsprofilierung umfasst das Hybridisieren von markierten cDNA- oder RNA-Proben mit einem etablierten Array von Oligonukleotid-Sonden. Die Signalintensität jeder Sonde korreliert direkt mit der Häufigkeit der spezifischen mRNA, die in der Probe vorhanden ist, und bietet somit eine quantitative Messung der Genexpression.
Diese Technik ermöglicht es Forschern, die Genexpression unter verschiedenen Bedingungen, Behandlungen oder Entwicklungsstadien zu untersuchen. Dadurch werden tiefgreifende Einblicke in die molekularen Mechanismen gewonnen, die verschiedene biologische Prozesse antreiben. Das Anwendungsspektrum ist umfangreich und reicht von der Untersuchung von Krankheitsmechanismen bis hin zur Erforschung der Entwicklungsbiologie.
Einführung in den Gene Expression Microarray Service
Die Genexpressions-Mikroarray-Technologie bietet einen robusten Ansatz, um das Transkriptom - das gesamte Set von RNA-Molekülen, die von einem Organismus unter bestimmten Bedingungen exprimiert werden - zu analysieren und zu quantifizieren. Zu den führenden Plattformen für die Genexpressionsprofilierung gehören die Affymetrix GeneChip- und Illumina BeadArray-Systeme. Diese Plattformen ermöglichen es Forschern, Genexpressionsmuster auf genomischer Ebene zu bewerten.
Affymetrix GeneChip
Das Affymetrix GeneChip-System nutzt eine Reihe von Mikroarrays, die für die genomweite Expressionsprofilierung entwickelt wurden. Das Clariom S Array ermöglicht eine umfassende Analyse, die die Expression von mRNA, lncRNA und miRNA umfasst. Das Clariom D Array hingegen bietet eine detailliertere Untersuchung, die sich auf alternative Isoformen und genomische Deletionen konzentriert. Dank seiner hochauflösenden Fähigkeiten sind die Affymetrix-Mikroarrays besonders gut geeignet, um komplexe Genexpressionsprofile zu entschlüsseln.
Illumina BeadArray
Im Gegensatz dazu verwendet die Illumina BeadArray-Plattform Perlen, die mit Oligonukleotid-Sonden beschichtet sind, um die Genexpression zu erfassen und zu messen. Dieses anpassungsfähige System unterstützt sowohl maßgeschneiderte als auch vordesignte Arrays und bedient ein Spektrum von Forschungsanwendungen – von moderaten Studien bis hin zu umfangreichen genomischen Erhebungen. Die Illumina-Technologie ist bekannt für ihre hohe Durchsatzrate und außergewöhnliche Reproduzierbarkeit, was sie zu einem wichtigen Asset für großangelegte Forschungsinitiativen macht.
Die Erstellung umfassender Expressionsprofile ist entscheidend für das Verständnis sowohl normaler biologischer Funktionen als auch von Krankheitsmechanismen. Bei CD Genomics setzen wir hochauflösende Array-Scans und Automatisierung ein, um die Effizienz von Genexpressionsstudien erheblich zu steigern. Unser Fachwissen umfasst die Verarbeitung von Mikroarrays verschiedener Anbieter, darunter Illumina Infinium, Affymetrix GeneChip und Agilent SureSelect, um nur einige zu nennen, und das alles unter Verwendung standardisierter Folienformate.
Diese vielfältigen Mikroarray-Designs sind sorgfältig auf der Grundlage des neuesten Genom-Inhalts entwickelt worden und ermöglichen die Erstellung von genomweiten Expressionsprofilen für Menschen, Modellorganismen, Pflanzen und Tiere. Um unterschiedlichen Forschungszielen gerecht zu werden, bieten wir eine umfassende Palette von Arrays an, die Analysen auf Ebene des gesamten Transkriptoms, von Genen, Exons und kurzen nicht-kodierenden RNAs (sncRNA) abdecken.
Unsere Arrays sind mit einem breiten Spektrum an Probenarten kompatibel und können niedrige RNA-Eingaben verarbeiten, was Flexibilität in verschiedenen experimentellen Setups gewährleistet. Darüber hinaus bieten wir sowohl Einzelproben-Array-Kartuschen als auch Mehrproben-Array-Platten an, um unterschiedlichen Durchsatzanforderungen gerecht zu werden und eine Vielzahl von Forschungsbedürfnissen zu bedienen.
Tabelle 1. Unser verfügbares Genexpressions-Mikroarray
| Menschen | Tiere | Pflanzen |
|
|
|
Anwendungen des Gene-Expression-Profiling-Mikroarray-Services
- Verstehen von KrankheitsmechanismenDurch die Untersuchung von Veränderungen in der Genexpression, Mikroarray kann die molekularen Grundlagen verschiedener Krankheiten aufdecken, einschließlich Krebs, Herz-Kreislauf-Erkrankungen und neurodegenerativen Erkrankungen. Solche Erkenntnisse sind entscheidend für die Entwicklung neuer therapeutischer Strategien.
- Biomarker identifizierenMikroarrays sind entscheidend für die Entdeckung von Biomarkern. Diese molekularen Signaturen sind unerlässlich für eine genaue Krankheitsdiagnose, die Bewertung der Prognose und die Überwachung von Therapieantworten.
- Entwicklungsprozesse erkundenForscher nutzen Genexpressionsprofile, um verschiedene Entwicklungs- und Differenzierungsstadien in verschiedenen Organismen zu untersuchen. Diese Forschung beleuchtet die komplexen biologischen Prozesse, die das Wachstum und die Reifung steuern.
- Bewertung von ArzneimittelreaktionenDurch die Nutzung von Mikroarrays können Wissenschaftler die Auswirkungen verschiedener Medikamente auf die Genexpression bewerten. Diese Anwendung ist besonders wertvoll in Arzneimittelentwicklung und im Bereich der personalisierten Medizin, wo das Verständnis individueller Reaktionen auf Behandlungen von größter Bedeutung ist.
- Umweltwirkungen untersuchenMikroarrays werden auch eingesetzt, um zu untersuchen, wie Umweltfaktoren wie Toxine und Ernährungsänderungen die Genexpression beeinflussen. Diese Forschung verbessert unser Verständnis der Wechselwirkungen zwischen externen Bedingungen und biologischen Systemen.
Vorteile des Gene-Expression-Profiling-Mikroarray-Services
- Genau und gleichzeitige Ausdrucksanalyse des gesamten Transkriptoms
- Flexible, maßgeschneiderte Lösungen für jedes Projekt
- Verfügbar für viele Arten
- Kosten-Nutzen-Analyse unter Verwendung des Standardkatalogs oder maßgeschneiderter Arrays
- Außergewöhnliches Studiendesign, Datenanalyseoptionen und Unterstützung
- Seelenfrieden mit vollständiger Probenverfolgung und Qualitätskontrolle
- Schnelle Lieferung von hochwertigen Ergebnissen durch automatisierte Probenverarbeitung
Genexpressionsprofilierung Mikroarray-Workflow
Der allgemeine Arbeitsablauf für die Genexpressions-Mikroarray ist unten skizziert. Wir haben drei gut anerkannte Genotypisierung Plattformen zusammen mit verschiedenen Arrays, die unterschiedliche Arten abdecken. Unser hochqualifiziertes Expertenteam führt das Qualitätsmanagement durch und befolgt jeden Schritt, um zuverlässige und unvoreingenommene Ergebnisse zu gewährleisten.

Dienstspezifikationen
Musteranforderungen
|
|
 Klicken |
Sequenzierungsstrategie
|
 |
Bioinformatikanalyse Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an:
|
Analyse-Pipeline
Liefergegenstände
- Rohdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
Unser Full-Service umfasst den gesamten Prozess, von der anfänglichen Qualitätskontrolle der Proben bis hin zur umfassenden Datenanalyse. Darüber hinaus bietet CD Genomics die Flexibilität, maßgeschneiderte Lösungen zu entwerfen. Mikroarray auf die spezifischen Projektanforderungen zugeschnitten. Sollten Sie weitere Anforderungen oder Fragen haben, zögern Sie bitte nicht, uns zu kontaktieren.
Demo-Ergebnisse
Teilweise Ergebnisse sind unten aufgeführt:
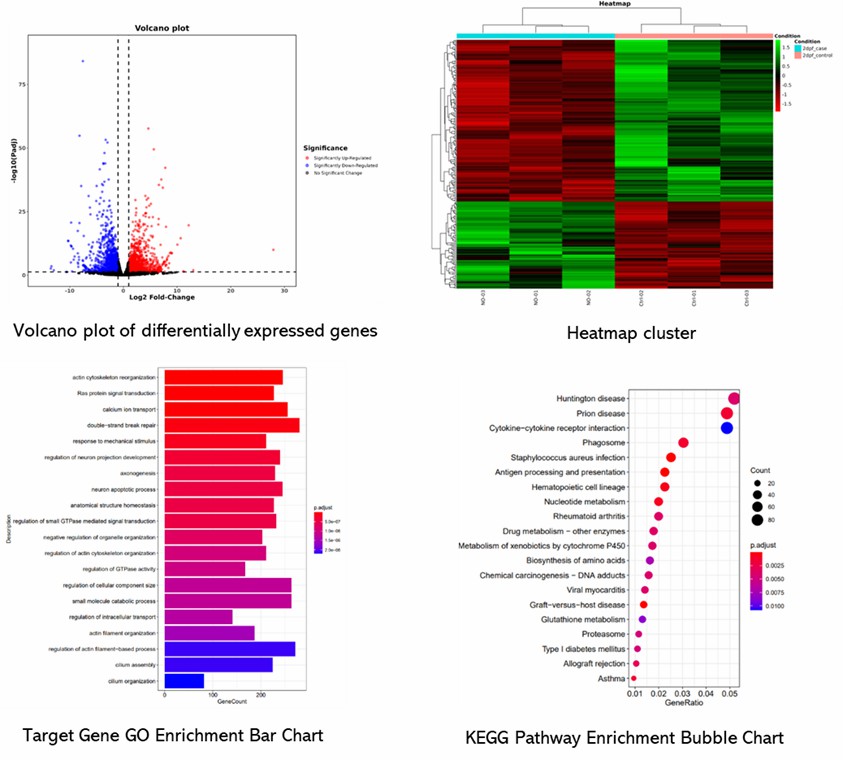
Häufig gestellte Fragen zum Mikromatrix-Service für die Genexpressionsprofilierung
1. Wie vergleicht sich die Genexpressionsprofilierung mit RNA-Sequenzierung (RNA-seq)?
Genexpressionsmikroarrays bieten einen gut etablierten, kosteneffektiven Ansatz zur Analyse der Genexpression über viele Gene hinweg. Im Gegensatz dazu bietet RNA-seq einen umfassenderen Datensatz, mit der zusätzlichen Fähigkeit, neuartige Transkripte und Isoformen zu erkennen. Dennoch sind die Kosten und die Komplexität von RNA-seq im Vergleich zur Mikroarray-Analyse höher.
2. Was sind die Hauptprobleme, die mit Genexpressions-Mikroarrays verbunden sind?
Eine große Herausforderung bei Genexpressions-Mikroarrays ist die Spezifität der Sonden, die die Genauigkeit der Ergebnisse beeinträchtigen kann. Darüber hinaus ist eine sorgfältige Probenvorbereitung entscheidend, um technische Artefakte zu vermeiden, die die Datenqualität und -integrität gefährden könnten.
3. Können Mikroarray-Daten mit anderen Omics-Daten integriert werden?
Ja, Mikroarray Daten können nahtlos mit anderen Omics-Datensätzen, wie Proteomik und Metabolomik, integriert werden. Diese Integration ermöglicht eine ganzheitlichere und umfassendere Sicht auf biologische Systeme und verbessert unser Verständnis ihrer Komplexität und Vernetztheit.
4. Wie können wir sicherstellen, dass die Proben, die für die Hybridisierung verwendet werden, angemessen an die Sonden auf dem Mikroarray binden? Gibt es Bedenken hinsichtlich einer Übersättigung?
Um sicherzustellen, dass die markierten Proben, die für die Hybridisierung verwendet werden, angemessen an die Sonden auf dem Mikroarray binden, ist es wichtig, die relativen Mengen von Sonden und markierten Proben zu berücksichtigen. Typischerweise übersteigt die Menge der entsprechenden Hybridisierungs-Sonden, die auf dem Mikroarray fixiert sind, bei weitem die Menge der markierten Proben, was Bedenken hinsichtlich einer möglichen Sättigung der Sonden verringert. Bei Dual-Channel-Mikroarrays können die fixierten Sonden auf dem Chip ausreichend gewährleisten, dass beide Arten von fluoreszenzmarkierten Proben vollständig an ihre jeweiligen fixierten Sonden binden.
5. Welche RNA-Menge wird benötigt, um eine Probe mit Genexpressions-Mikroarrays zu analysieren?
Für die Analyse einer Probe mit Genexpressions-Mikroarrays haben verschiedene Plattformen spezifische RNA-Mengenanforderungen. Agilent-Expressions-Mikroarrays benötigen eine minimale Gesamt-RNA-Menge von 200 ng für sowohl eukaryotische als auch prokaryotische Organismen. Im Gegensatz dazu erfordern Affymetrix-Mikroarrays mindestens 250 ng Gesamt-RNA.
Gene-Expressions-Profilierung Mikroarray-Dienst Fallstudien
Vergleich von RNA-Seq und Mikroarray-Gene-Expressionsplattformen zur toxikogenomischen Bewertung der Leber aus kurzzeitigen Ratten-Toxizitätsstudien
Zeitschrift: Frontiers in Genetics
Impactfaktor: 4,772
Veröffentlicht: 22. Januar 2019
Hintergrund
Toxikogenomik verwendet die Genexpressionsprofilierung zur Bewertung von Toxizität, zunächst mit Mikroarrays und zunehmend auch mit RNA-SeqRNA-Seq bietet eine detailliertere Ansicht, indem es das gesamte Transkriptom sequenziert, hat jedoch größere Datendateien und eine komplexere Analyse im Vergleich zu Mikroarrays. Trotz seiner Vorteile gibt es nur begrenzte Vergleiche zwischen RNA-Seq und Mikroarrays in Toxizitätsstudien, was einen weiteren Forschungsbedarf zur relativen Wirksamkeit bekräftigt.
Materialien & Methoden
Probenvorbereitung
- Männliche Ratten
- Schnellgefrorenes Leber
- RNA-Extraktion
Methode
- Hauptkomponentenanalyse (PCA)
- Genexpressionsanalyse
- Statistische Analysen
Ergebnisse
Histopathologie: Die Verabreichung von ANIT, MDA und CCl4 führte zu erwarteter Hepatotoxizität, gekennzeichnet durch erhöhte Serumleberenzyme und entsprechende histopathologische Veränderungen der Leber. APAP und DCLF zeigten keine signifikante Leberpathologie.
Hauptkomponentenanalyse (PCA): Die PCA von RNA-Seq- und Mikroarray-Daten zeigte eine klare Trennung der ANIT-, MDA- und CCl4-Proben von den Kontrollen, während die APAP-behandelten Proben sich nicht trennten, was auf minimale Veränderungen der Genexpression hinweist.
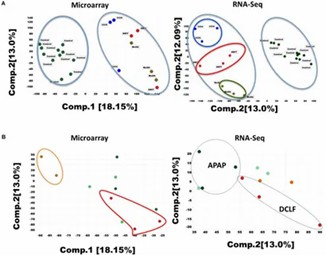 Abbildung 1. (A) Hauptkomponentenanalyse (PCA) des RNA-seq- und Mikroarray-Datensatzes für 26 Leberproben.
Abbildung 1. (A) Hauptkomponentenanalyse (PCA) des RNA-seq- und Mikroarray-Datensatzes für 26 Leberproben.
Absolute Gene-Expressionsübereinstimmung: Die Spearman-Korrelationskoeffizienten zeigten eine hohe Übereinstimmung zwischen den RNA-Seq- und Mikroarray-Plattformen für ANIT, MDA und CCl4. Allerdings wiesen APAP und DCLF schwächere Korrelationen auf, was auf die Empfindlichkeit von RNA-Seq gegenüber niedrig exprimierten Genen hinweist.
DEGs Übereinstimmung: RNA-Seq identifizierte mehr DEGs im Vergleich zu Mikroarrays und zeigte eine höhere Sensitivität sowie eine bessere Leistung bei der Erfassung von Veränderungen der Genexpression, insbesondere bei niedrig exprimierten und stark dysregulierten Genen.
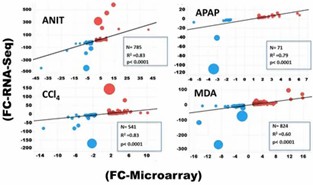 Abbildung 2. Spearman-Korrelationsdiagramm für DEGs, die durch RNA-Seq und Mikroarray bestimmt wurden.
Abbildung 2. Spearman-Korrelationsdiagramm für DEGs, die durch RNA-Seq und Mikroarray bestimmt wurden.
Einzigartige DEG-Erkennung: RNA-Seq identifizierte für alle getesteten Hepatotoxine mindestens 10-mal mehr einzigartige protein-codierende DEGs im Vergleich zu Mikroarrays, was die größere Sensitivität von RNA-Seq hervorhebt.
 Abbildung 3. Hierarchisch gruppierte Gene (Spalten) und Proben (Zeilen) mit Dendrogrammen und Clustern (blau gefärbte Balken).
Abbildung 3. Hierarchisch gruppierte Gene (Spalten) und Proben (Zeilen) mit Dendrogrammen und Clustern (blau gefärbte Balken).
Kanonische Wege: Sowohl RNA-Seq als auch Mikroarrays identifizierten ähnliche lebertoxizitätsbezogene Wege, jedoch entdeckte RNA-Seq zusätzliche Wege. Beide Plattformen zeigten unterschiedliche leberassoziierte Wege für jeden Hepatotoxikanten.
Upstream-Regulatoren: Beide Plattformen identifizierten überlappende und einzigartige upstream-Regulatoren, wobei RNA-Seq zusätzliche Regulatoren offenbarte, die durch Mikroarrays nicht gefunden wurden.
Nicht-kodierende DEGs: RNA-Seq hat auch eine Vielzahl von nicht-kodierenden RNAs nachgewiesen, wobei in mehreren Kategorien eine signifikante Regulation beobachtet wurde. Differenziell exprimierte lncRNAs zeigten potenzielle Korrelationen mit nahegelegenen protein-kodierenden Genen und lieferten Einblicke in ihre biologischen Rollen.
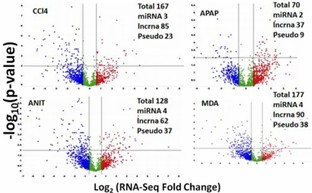 Abbildung 4. Vulkan-Diagramm, das spezifische nicht-kodierende differentielle exprimierte Gene (DEGs) aus RNA-Seq zusammenfasst.
Abbildung 4. Vulkan-Diagramm, das spezifische nicht-kodierende differentielle exprimierte Gene (DEGs) aus RNA-Seq zusammenfasst.
Fazit
RNA-Seq übertrifft Mikroarray-Analysen bei der Erkennung von Lebertoxizität, indem es mehr differentielle exprimierte Gene (DEGs) und relevante Signalwege identifiziert, einschließlich nicht-kodierender Gene. Seine höhere Sensitivität und Genauigkeit machen es zu einem wertvollen Werkzeug, obwohl verbesserte RNA-Seq-Datenbanken für eine bessere Datenintegration erforderlich sind.
Referenz
- Rao MS, Van Vleet TR, Ciurlionis R, et al. Vergleich von RNA-Seq- und Mikroarray-Genexpressionsplattformen für die toxikogenomische Bewertung der Leber aus kurzfristigen Ratten-Toxizitätsstudien. Grenzen in der Genetik. 2019, 9:636.
Verwandte Veröffentlichungen
Hier sind einige Veröffentlichungen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Einsatz von Biostimulanzien zur Minderung von Wasserstress in zwei Hartweizen (Triticum durum Desf.) Genotypen mit unterschiedlicher Trockenheitstoleranz
Zeitschrift: Pflanzenstress
Jahr: 2024
Die Restriktions-Modifikationssysteme von Clostridium carboxidivorans P7
Zeitschrift: Mikroorganismen
Jahr: 2023
Im Land der Blinden: Außergewöhnliche subterranne Spezialisierung von kryptischen troglobitischen Spinnen der Gattung Tegenaria (Araneae: Agelenidae) in Israel
Zeitschrift: Molekulare Phylogenetik und Evolution
Jahr: 2023
Genetische Modifikatoren des oralen Nikotinkonsums bei Chrna5-Nullmutantenmäusen
Zeitschrift: Front. Psychiatrie
Jahr: 2021
Eine hochdichte genetische Verknüpfungskarte und QTL-Identifizierung für Wachstumsmerkmale bei Dunkelkob (Argyrosomus japonicus)
Zeitschrift: Aquakultur
Jahr: 2024
Genomische und chemische Beweise für lokale Anpassung an die Resistenz gegenüber verschiedenen Herbivoren in Datura stramonium
Zeitschrift: Evolution
Jahr: 2020
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


