
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
MeDIP‑Seq / hMeDIP‑Seq (DNA-Methylierungs- und Hydroxymethylierungsprofilierung)
CD Genomics bietet MeDIP-seq für die genomweite Methylierungsprofilierung und hMeDIP-seq an, um selektiv 5-Hydroxymethylcytosin (5hmC) zu erfassen, was eine parallele Analyse mehrerer Cytosin-Modifikationen ermöglicht.
Einführung in MeDIP-Seq und hMeDIP-Seq
DNA-Methylierung bezieht sich auf die postreplikative Erhaltung oder die de novo Hinzufügung einer Methylgruppe an der Kohlenstoff-5-Position des Cytosin-Pyrimidinrings durch DNA-Methyltransferasen. Bei Säugetieren tritt DNA-Methylierung typischerweise in CG (mCG) und non-CG (mCHG oder mCHH, gemeinsam als mC bezeichnet) Kontexten auf. mCG allein macht etwa 60–80% aller CG-Dinukleotide aus. Neben 5-Methylcytosin (5mC) wurden mehrere Cytosinmodifikationen identifiziert, darunter 5-Hydroxymethylcytosin (5hmC), 5-Formylcytosin und 5-Carboxylcytosin. Unter ihnen ist 5hmC strukturell ähnlich zu 5mC, jedoch viel seltener und wird durch die Oxidation von 5mC durch die TET-Familie der Dioxygenasen erzeugt.
DNA-Methylierung und Hydroxymethylierung sind beide entscheidende epigenetische Marker, die an der Genregulation, embryonalen Entwicklung, zellulären Differenzierung und Genomstabilität beteiligt sind. Die genomweite Analyse sowohl von 5mC als auch von 5hmC bietet wichtige Einblicke in die epigenetische Dynamik, insbesondere in Studien zur Entwicklung und Stressreaktion.
MeDIP‑seq kombiniert Immunpräzipitation mit Hochdurchsatz-Sequenzierung um 5mC im gesamten Genom anzureichern und zu profilieren. Es nutzt Anti-5mC-Antikörper, um DNA-Fragmente, die methylierte Cytosine (in mCG- und mCH-Kontexten) enthalten, selektiv zu erfassen. Diese angereicherten Fragmente werden sequenziert, und ihre Leseverteilung kann verwendet werden, um relative Methylierungsniveaus zu schätzen.
Um 5hmC zu unterscheiden und spezifisch zu profilieren, bietet CD Genomics auch hMeDIP‑seq an, das 5hmC-spezifische Antikörper verwendet, um hydroxymethylierte DNA-Fragmente anzureichern. Dieser ergänzende Ansatz ermöglicht es Forschern, 5mC- und 5hmC-Modifikationen parallel separat zu profilieren, wodurch die Auflösung der epigenetischen Kartierung über verschiedene Probenarten hinweg verbessert wird. Sowohl MeDIP‑seq als auch hMeDIP‑seq sind nicht-destruktive, umwandlungsfreie Methoden, die nicht auf Bisulfitbehandlung oder uracil-tolerante Polymerasen angewiesen sind und mit Proben mit niedrigem Input kompatibel sind.
Vorteile von MeDIP-Seq / hMeDIP-Seq
- Kann mC, mCG oder hmC anvisieren.
- Ganzes Genom oder bestimmte Interessensregionen
- Nahezu unvoreingenommen und Hypothese
- Entdeckung epigenetischer Biomarker
- Einzel-Nukleotid-Auflösung und Kosten-Effizienz
- Niedriger DNA-Eingangsbetrag
Anwendungen von MeDIP-Seq / hMeDIP-Seq
- Epigenetische Heterogenität
- Umwelt und Epigenetik
- Regulation der genetischen Expression
- Krankheitsforschung
- Genetische Prägung
- Embryonale Entwicklung
- Erkennung von DNA-Methylierungs-angereicherten Regionen in Krankheitsproben und Ableitung von Kandidatengen-Sets, deren Expression in den Proben unterdrückt ist.
- Überwachung der dynamischen DNA-Methylierungsmuster in verschiedenen Phasen des Auftretens und der Entwicklung von Krankheiten, insbesondere an Läsionsstellen, um epigenetische Marker zu identifizieren, die helfen, das Ausmaß des Krankheitsfortschritts zu definieren.
- Vergleichende Analyse der Unterschiede in der Signal- und Standortverteilung von DNA-Methylierungsregionen zwischen Krankheits- und Normalproben, Identifizierung von krankheitsspezifischen DNA-Methylierungsregionen und Beobachtung der Gene in der Umgebung dieser Regionen, um die Liste der Kandidatengene, die mit der Krankheit in Verbindung stehen, einzugrenzen.
- Kartierung der Hydroxymethylierung (5hmC) Landschaften in Geweben, Zell-Differenzierungs- oder Stressreaktionsmodellen unter Verwendung von hMeDIP-seq.
MeDIP-Seq / hMeDIP-Seq Arbeitsablauf
Der allgemeine Arbeitsablauf für MeDIP-Sequenzierung ist unten skizziert. Kurz gesagt, wird die extrahierte DNA fragmentiert, denaturiert, mit Adaptern ligiert und mit dem Antikörper, der gegen 5-Methylcytosin (für MeDIP-seq) oder 5-Hydroxymethylcytosin (für hMeDIP-seq) gerichtet ist, gefangen, gefolgt von der Bibliotheksvorbereitung und Sequenzierung auf Illumina-Plattformen.

Dienstspezifikation
Musteranforderungen
|
|
 Klicken |
Sequenzierungsstrategien
|
 |
Datenanalyse Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an:
|
Analyse-Pipeline

Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details in MeDIP-Seq / hMeDIP-Seq für Ihre Schreibanpassung.
Mit professionellen bioinformatischen Fähigkeiten bietet CD Genomics einen hochwertigen MeDIP-Seq-Service als umfassenden, genomweiten epigenetischen Service an, um unterschiedlich methylierten Regionen zu identifizieren und letztendlich die epigenetische Forschung zu beschleunigen. Wenn Sie zusätzliche Anforderungen oder Fragen haben, zögern Sie bitte nicht, sich zu melden. kontaktieren Sie uns.
Referenz:
- Taiwo O, Wilson G A, Morris T, u. a.Methylom-Analyse mit MeDIP-seq bei niedrigen DNA-Konzentrationen. Naturprotokolle, 2012, 7(4): 617.
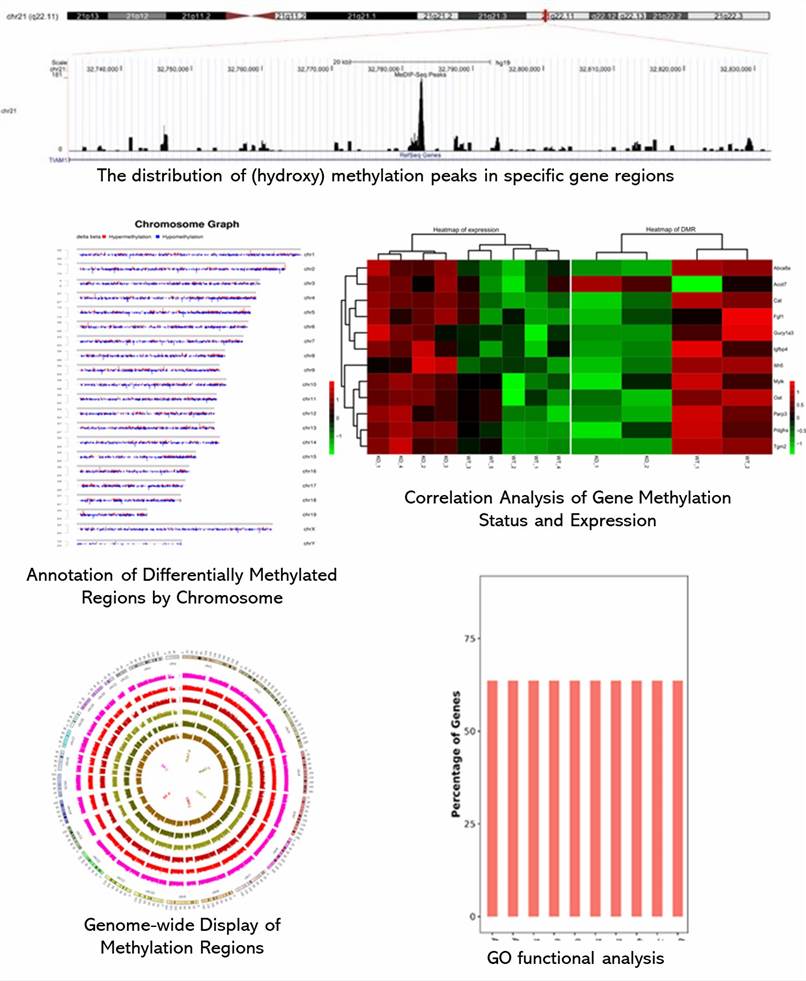
Demo-Ergebnisse
Teilweise Ergebnisse sind unten aufgeführt:
MeDIP‑Seq / hMeDIP‑Seq häufig gestellte Fragen
1. Im Vergleich zu anderen Methoden, welche Vorteile bietet die MeDIP-Sequenzierung?
Derzeit kann der sequenzierungsbasierte Ansatz zur Methylom-Analyse in bisulfidkonversionsbasierte und anreicherungsbasierte Ansätze unterteilt werden. Zu den bisulfidkonversionsbasierten Methoden gehören Whole-Genome oder gezielt Bisulfit-Sequenzierung oder reduzierte Repräsentation Bisulfit-Sequenzierung (RRBS)Obwohl Methoden, die auf der Bisulfit-Konversion basieren, als der Goldstandard der DNA-Methylierungsanalyse mit Einzelbasenauflösung gelten, können sie zwischen mC und hmC nicht unterscheiden. Darüber hinaus, Whole-Genome-Bisulfid-Sequenzierung ist teuer für Anwendungen mit großen Stichprobengrößen und von kleineren Forschungsgruppen. Und RRBS kann nur eine begrenzte Genomabdeckung (5-10%) bieten und konzentriert sich auf CpG-Inseln und Promotorregionen.
Neben MeDIP-Sequenzierung umfassen anreicherungsbasierte Technologien auch MBD-seq, das die methylbindenden Proteine MBD2 und MDB3L1 verwendet, sowie methylCap-seq, das die methylbindende Domäne von MECP2 zur Methylfängung nutzt. MBD-seq und methylCap-seq sind jedoch auf die Analyse von mCG beschränkt, und Protokolle für das gesamte Genom erfordern oft hohe Konzentrationen an genomischer DNA (mehr als 1.000 ng). Daher ist MeDIP eine vielseitige, genaue und kostspielige Methode mit einem geringen DNA-Eingang und ist auf eine breite Palette von Proben und Studien anwendbar.
2. Was sind die Anforderungen an Proben für MeDIP-Sequenzierung?
MeDIP-Sequenzierungsproben benötigen ein Referenzgenom oder Sequenzen für die Ausrichtung, und die zusammengefassten Ergebnisse beeinflussen direkt die Genauigkeit der Datenanalyse. Ein Referenzgenom von eng verwandten Arten oder zusammengefasste Ergebnisse aus dem Transkriptom können ebenfalls verwendet werden, obwohl dabei teilweise Methylierungsinformationen verloren gehen.
3. Welche Faktoren können die Ergebnisse von MeDIP-seq beeinflussen?
Jeder Prozess, der an MeDIP-seq beteiligt ist, kann die Ergebnisse beeinflussen, insbesondere die Immunpräzipitation und PCR. Die Immunpräzipitation ist der Prozess zur Anreicherung von methylierte Regionen, und die Reaktionsbedingungen können die Anreicherungsresultate beeinflussen. Wenn nicht genügend DNA recycelt wird, wird PCR verwendet, um sie zu vergrößern, was die Ergebnisse verzerren kann. Darüber hinaus sollte Kontamination während des gesamten Prozesses vermieden werden.
MeDIP‑Seq / hMeDIP‑Seq Fallstudien
Epigenetische Veränderungen bei TRAMP-Mäusen: Epigenom-DNA-Methylierungsprofilierung mittels MeDIP-seq
Zeitschrift: Zellen & Biowissenschaften
Veröffentlicht: 12. Januar 2018
Zusammenfassung
Die Autoren profilierten das Methylom des Mausprostatakrebsmodells (TRAMP) und analysierten die Wechselwirkungen zwischen den gezielten Genen und den damit verbundenen funktionalen Wegen. Durch die Nutzung von MeDIP-Sequenzierung analysierten sie DNA-Methylierung Profile und führten relevante informatische Analysen durch, die Strategien für Präventions- und Behandlungsansätze bei Prostatakrebs bieten könnten.
Materialien & Methoden
- TRAMP-Mäuse und C57BL/6-Mäuse
- Genomische DNA-Extraktion
- MeDIP-seq
- Illumina HiSeq2000
- RT-PCR
- Methylierungs-spezifische PCR
- Ausrichtung an dem Referenz-Mausgenom
- Kanonische Wege
- Krankheiten und Funktions- sowie Netzwerkanalysen
Ergebnisse
1. Vergleich der MeDIP-seq-Ergebnisse
Insgesamt zeigten 2147 Gene zwischen TRAMP- und Kontrolltieren eine signifikante Veränderung in den methylierten Peaks. Im Vergleich zur Kontrolle wurden eine signifikant erhöhte Methylierung von 1042 Genen und eine signifikant verringerte Methylierung von 1105 Genen bei TRAMP beobachtet. Vier interessante Gene, DYNC1I1, SLC1A4, XRCC6BP1 und TTR, wurden mit IGV analysiert. TRAMP-Mäuse zeigten ein erhöhtes Methylierungsverhältnis von DYNC111 und SLC1A4 sowie ein verringertes Methylierungsverhältnis von TTR und XRCC6BP1, was mit den MeDIP-seq-Ergebnissen übereinstimmte.
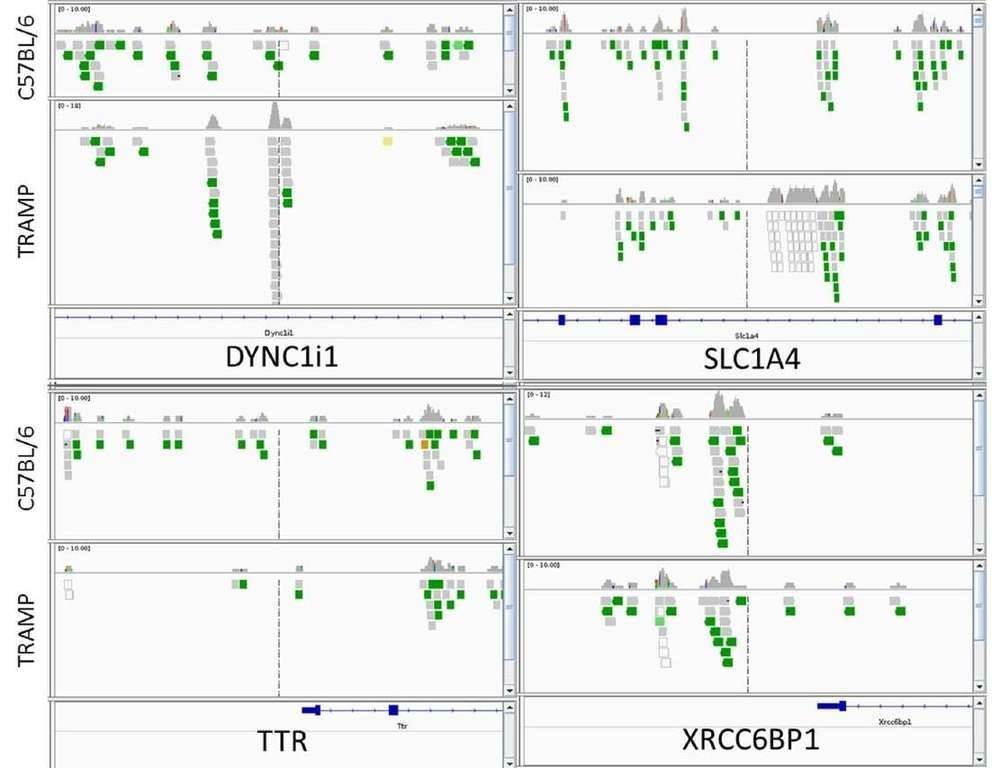 Abbildung 1. Integrative Genomics Viewer Visualisierung der Verteilung der ausgerichteten Reads gegen das Referenzgenom für vier Zielgene: DYNC1I1, SLC1A4, XRCC6BP1 und TTR.
Abbildung 1. Integrative Genomics Viewer Visualisierung der Verteilung der ausgerichteten Reads gegen das Referenzgenom für vier Zielgene: DYNC1I1, SLC1A4, XRCC6BP1 und TTR.
2. qPCR-Validierung der ausgewählten Genexpression
Die Expressionsniveaus von CRYZ, DYNC1I1, HNMT, SLC1A4 und TTR wurden sowohl in der TRAMP- als auch in der Wildtyp-Gruppe gemessen (Abbildung 2). Unter ihnen war die TTR-Expression um das 9,05-Fache im Vergleich zum Wildtyp erhöht. Zudem waren die Expressionsniveaus von TTR in Prostatakrebsgewebe signifikant höher als in normalem Gewebe und in Gewebe mit benigner Prostatahyperplasie. Darüber hinaus fanden sie eine verringerte Methylierung im Promotorbereich von TTR, jedoch eine erhöhte Genexpression. Im Gegensatz dazu kann die DNA-Methylierung im Genkörper oder stromabwärts einer reziproken Beziehung folgen oder auch nicht.
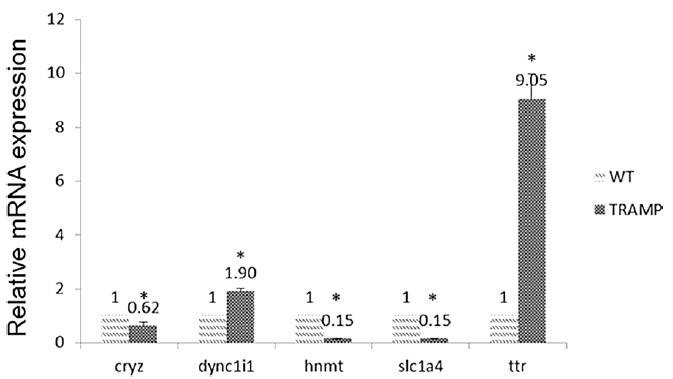 Abbildung 2. Vergleich der miRNA-Expression von CRYZ, DYNC1I1, HNMT, SLC1A4 und TTR zwischen Proben von Prostata des Wildtyps und TRAMP-Mäusen.
Abbildung 2. Vergleich der miRNA-Expression von CRYZ, DYNC1I1, HNMT, SLC1A4 und TTR zwischen Proben von Prostata des Wildtyps und TRAMP-Mäusen.
3. Kanonischer Weg, Krankheiten und Funktionen sowie Netzwerkanalysen.
Die 2147 Gene mit signifikanten Veränderungen in der Methylierung wurden analysiert. Die krebsbezogenen Netzwerke machten den Großteil aus (Tabelle 2), was darauf hindeutet, dass der Unterschied zwischen TRAMP und Kontrolle in der Organentwicklung und der Krebsentwicklung lag. Die am stärksten assoziierten Krankheiten, Krebs, gastrointestinale Erkrankungen, Organismusanomalien, Erkrankungen des Fortpflanzungssystems und dermatologische Erkrankungen, wurden unter den fünf wichtigsten eingestuft (Abbildung 3).
Tabelle 1. Die zehn wichtigsten veränderten kanonischen Signalwegen.
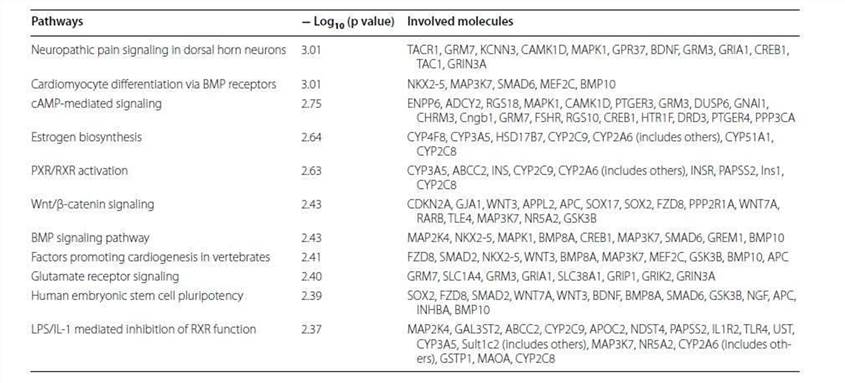
Tabelle 2. Top analysierte Netzwerke.
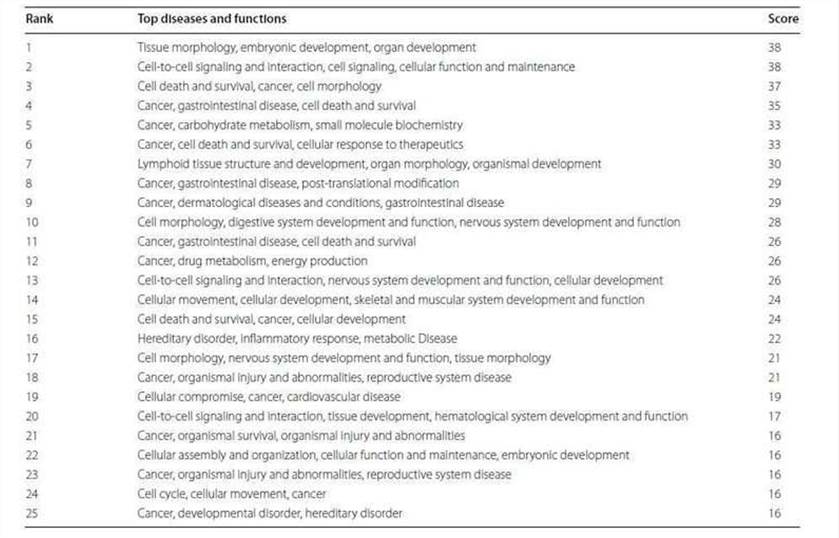
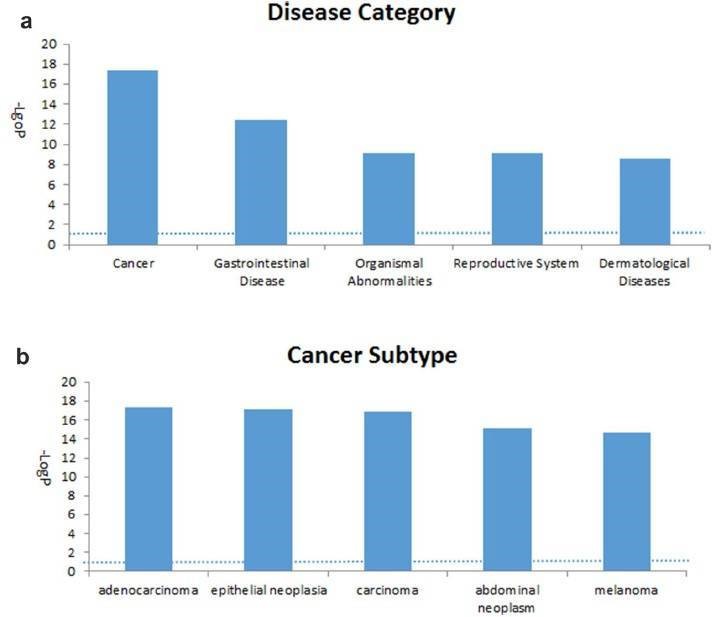 Abbildung 3. Die fünf wichtigsten assoziierten Krankheitskategorien (a) und die fünf wichtigsten Krebsuntertypen (b), die analysiert wurden.
Abbildung 3. Die fünf wichtigsten assoziierten Krankheitskategorien (a) und die fünf wichtigsten Krebsuntertypen (b), die analysiert wurden.
Diskussion
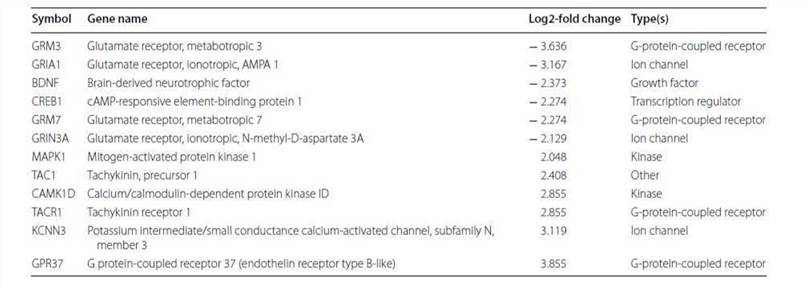
Die Analyse des kanonischen Weges würde Informationen für die Entwicklung neuer therapeutischer Ziele liefern. Wie in Abbildung 4 gezeigt, waren die Gene mit signifikanter Methylierung im wichtigsten kanonischen Weg der neuropathische Schmerzsignalweg, was mit der früheren Erkenntnis übereinstimmt, dass die häufigste Malignität bei TRAMP neuroendokrinen Ursprungs ist. Tabelle 3 listet die Gene auf, die an diesem Weg beteiligt sind und eine veränderte Methylierung aufwiesen. CREB wurde als eng mit Zellproliferation, Differenzierung und adaptiven Reaktionen im neuronalen System verbunden festgestellt, und die Methylierung des CREB1-Gens wurde bei TRAMP um 2,274 verringert. Darüber hinaus wurde auch festgestellt, dass CREB andere Karzinogenese-Wege reguliert. All diese Daten deuten darauf hin, dass CREB eng mit der Krebstherapie verbunden ist und eine neue Strategie zur Prävention und Therapie von Prostatakrebs darstellen könnte.
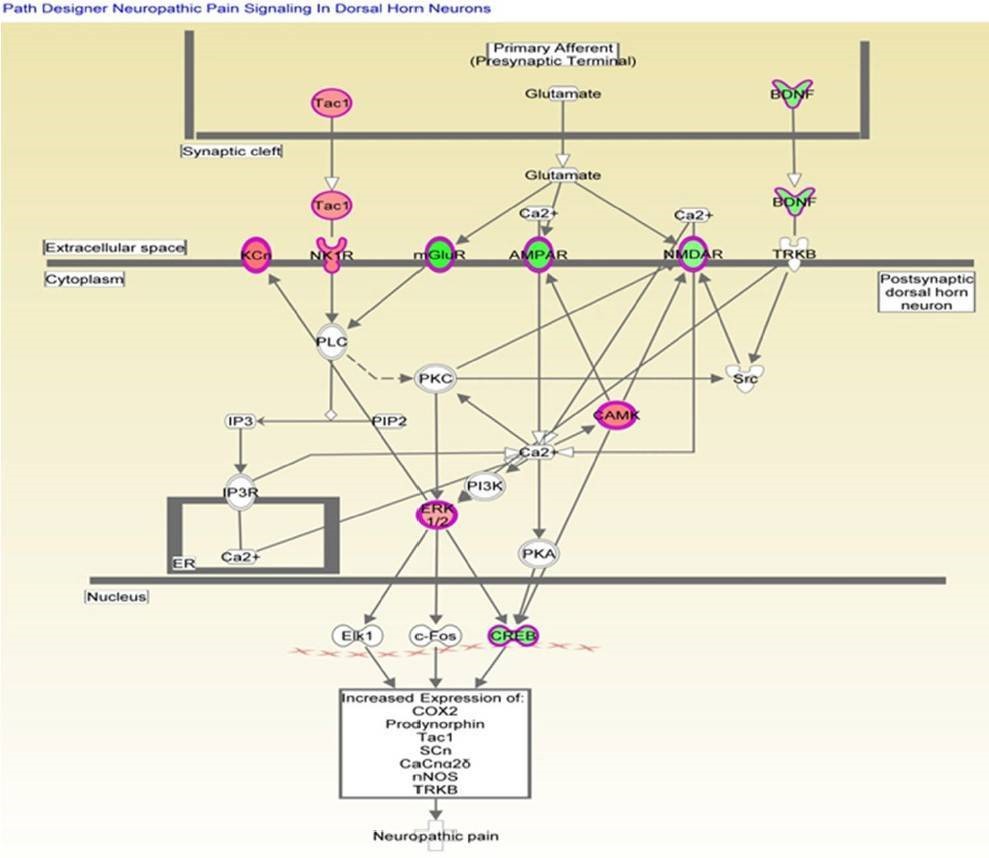 Abbildung 4. Gene, die dem kanonischen Signalweg für neuropathische Schmerzen zugeordnet sind.
Abbildung 4. Gene, die dem kanonischen Signalweg für neuropathische Schmerzen zugeordnet sind.
Tabelle 3. Veränderte Methylierungsgene, die dem Signalweg der neuropathischen Schmerzen zugeordnet sind.
Referenz:
- Li W, Huang Y, Sargsyan D, u. a.Epigenetische Veränderungen bei TRAMP-Mäusen: Epigenom-DNA-Methylierungsprofilierung mittels MeDIP-seq. Zell- und Biowissenschaften, 2018, 8(1): 3.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Die Spaltung von Phagen-DNA durch Restriktionsendonukleasen ermöglicht die Wiederbelebung aus der durch Cas13 induzierten bakteriellen Dormanz.
Zeitschrift: Nature Mikrobiologie
Jahr: 2023
IL-4 fördert die Erschöpfung von CD8.+ CART-Zellen
Journal: Naturkommunikationen
Jahr: 2024
Fettreiche Diäten während der Schwangerschaft verursachen Veränderungen der DNA-Methylierung und Proteinexpression im Pankreasgewebe des Nachwuchses: Ein Multi-Omics-Ansatz
Internationale Zeitschrift für Molekulare Wissenschaften
Jahr: 2024
KMT2A assoziiert mit dem PHF5A-PHF14-HMG20A-RAI1 Subkomplex in Stammzellen des Pankreaskarzinoms und reguliert epigenetisch deren Eigenschaften.
Journal: Naturkommunikationen
Jahr: 2023
Krebsassoziierte DNA-Hypermethylierung von Polycomb-Zielen erfordert die doppelte Erkennung von Histon H2AK119-Ubiquitinierung und der sauren Tasche des Nukleosoms durch DNMT3A.
Journal: Wissenschaftliche Fortschritte
Jahr: 2024
Genomisches Imprinting-ähnliches monoalleles väterliches Ausdrucksmuster bestimmt das Geschlecht von Kanalkatzenfischen.
Journal: Wissenschaftliche Fortschritte
Jahr: 2022
Mehr ansehen Artikel, die von unseren Kunden veröffentlicht wurden.


