
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
MeRIP-Sequenzierung (m6A-Analyse)
CD Genomics bietet die transkriptomweite Analyse der Position von m6A in mRNA-Transkripten und anderen RNAs durch methylierte RNA-Immunpräzipitation-Sequenzierung (MeRIP-seq) an. MeRIP-seq verwendet einen Antikörper, der spezifisch N6-Methyladenosin (m6A) in RNA nachweist und charakterisiert. Da die 3'UTR von mRNA eine wichtige Rolle bei der Stabilität, Lokalisation und Translation von mRNA spielt und die Bindung von RNA-regulatorischen Proteinen an diese Regionen beeinflussen kann, deutet eine Anreicherung von m6A in diesem Bereich darauf hin, dass m6A den RNA-Stoffwechsel und die Genexpression beeinflussen könnte. Die Hemmung der Enzyme, die an der RNA-Methylierung beteiligt sind, hat ebenfalls Hinweise auf die potenziellen biologischen Rollen dieser Modifikation geliefert. Daher kann die Identifizierung und Charakterisierung von modifizierten Basen in RNA zu einem besseren Verständnis biologischer Signalwege führen.
Was ist MeRIP-Sequenzierung?
N6-Methyladenosin (m6A), die häufigste Form der Methylierungsmodifikation innerhalb des mRNA-Sequenzen Eukaryoten haben die Fähigkeit, das eukaryotische Transkriptom funktionell zu regulieren, wodurch die Prozesse der mRNA-Spleißung, des nukleären Exports, der Lokalisation, der Translation und der Stabilität beeinflusst werden. Das Vorhandensein von m6A-RNA in verschiedenen lebenswichtigen zellulären Prozessen zeigt, dass die mRNA-Methylierung an zahlreichen biologischen Prozessen beteiligt ist, wie z.B. der Differenzierung von Stammzellen und dem zirkadianen Rhythmus, und sie ist an der Pathogenese verschiedener Krankheiten beteiligt, einschließlich, aber nicht beschränkt auf Karzinogenese, Fettleibigkeit und Unfruchtbarkeit.
Die primäre Technik für die Transkriptomforschung zu N6-Methyladenosin (m6A) ist MeRIP-seq (methylierte RNA-Immunpräzipitation-Sequenzierung). Das zugrunde liegende Prinzip dieser Methode besteht in der Verwendung von m6A-spezifischen Antikörpern, um RNA-Fragmente mit m6A-Modifikationen innerhalb der Zelle zu immunpräzipitieren, gefolgt von einer Hochdurchsatz-Sequenzierung dieser angereicherten RNA-Fragmente. In Kombination mit bioinformatischer Analyse kann eine umfassende systematische Forschung zu m6A-Modifikationen auf transkriptomweiter Ebene durchgeführt werden.
Vorteile unseres MeRIP-Sequenzierungsdienstes
- Hohe Flexibilität: In der Lage, hochmethylierten Fragmenten des Transkriptoms aus jeder Art direkt zu sequenzieren, ohne dass Informationen über die bekannte Genomsequenz erforderlich sind.
- Hohe Präzision: In der Lage, innerhalb eines Bereichs von 100-200 Nukleotiden um die tatsächliche Bindungsstelle präzise zu lokalisieren.
- Digitale Signale: Ermöglichen eine quantitative und sequenzielle Analyse direkt an methylierten Fragmenten, wodurch Kreuzreaktivität und Hintergrundrauschen, die mit traditionellen Chip-Hybridisierungsfluoreszenzanalogsignalen verbunden sind, beseitigt werden.
- Umfassende Plattform: MeRIP-Experiment—Bibliothekskonstruktion, Sequenzierungsanalyse—MeRIP qPCR-Validierung, die einen echten Rundum-Service bietet.
- Flexible Auswahl: Optionen für Bibliothekskonstruktionsmethoden und Fragmentgrößen.
- Gute Reproduzierbarkeit: Umfangreiche Projekterfahrung gewährleistet die experimentelle Reproduzierbarkeit, wobei vergleichende Analysen zwischen mehreren Proben durchgeführt werden.
Anwendung der MeRIP-Sequenzierung
- Verteilung der RNA-Methylierung in eukaryotischen Organismen;
- mRNA-Methylierung bei Tieren und Pflanzen und deren Reaktionsmechanismus auf Umweltstress;
- Mechanismen des Widerstands gegen mRNA-Abbau;
- Mechanismen, die der Tumorinitiierung und -proliferation zugrunde liegen;
- Biologische Funktionsstudien verschiedener Pflanzen;
- Krebsentstehung und Metastasierung; Krankheitsbeginn und -fortschritt;
- Zellendifferenzierung, embryonale Entwicklung und Stressreaktion.
MeRIP-Sequenzierungs-Workflow
CD Genomics führt den m6A-Mapping-Ansatz unter Verwendung von Immunpräzipitation mit m6A-spezifischen Antikörpern durch, gefolgt von NGS (MeRIP-Seq). Poly(A)-selektierte RNA wurde in 100-Nukleotid-lange Oligonukleotide (Input) fragmentiert und mit einem anti-m6A affin gereinigten Antikörper immunpräzipitiert, bevor sie der Illumina-Nächste-Generations-Sequenzierung unterzogen wurde. Da immunpräzipitierte RNA-Fragmente m6A an beliebiger Stelle entlang ihrer Länge enthalten können, erzeugen mehrere verschiedene m6A-haltige Fragmente überlappende Reads. m6A-Stellen wurden bioinformatisch vorhergesagt, indem ein Peak-Detektionsalgorithmus verwendet und nach der Anwesenheit einer Teilmenge von DRACH-Motiven in der Nähe des Punktes mit der höchsten Read-Abdeckung gesucht wurde.
 Abb. 1 MeRIP-Sequenzierungsprotokoll
Abb. 1 MeRIP-Sequenzierungsprotokoll
Dienstspezifikationen
Musteranforderungen
|
|
 |
Sequenzierung
|
 |
Bioinformatische Analyse
|
Analyse-Pipeline

Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details zur MeRIP-Sequenzierung für Ihre Schreibanpassung.
Die MeRIP-Sequenzierungskonferenz von CD Genomics konzentriert sich auf die Zusammenhänge zwischen Zellvariationen in Geweben und der Organfunktion und beleuchtet weiter die Ursprünge von Krankheiten. Wenn Sie zusätzliche Anforderungen oder Fragen haben, zögern Sie bitte nicht, uns zu kontaktieren.
Referenzen
- Dominissini D, et al. Topologie der m6A-RNA-Methylome von Mensch und Maus, aufgedeckt durch m6A-seq. Natur, 2012 Apr 29; 485(7397):201-6.
- Jesus D F D, et al. m6A mRNA-Methylierung reguliert die Biologie der menschlichen β-Zellen in physiologischen Zuständen und bei Typ-2-Diabetes. Naturstoffwechsel2019,1, 765–774.
Demo-Ergebnisse
 m6A mRNA-Methylierung reguliert die Biologie der menschlichen β-Zellen in physiologischen Zuständen und bei Typ-2-Diabetes. (Jesus et al., 2019)
m6A mRNA-Methylierung reguliert die Biologie der menschlichen β-Zellen in physiologischen Zuständen und bei Typ-2-Diabetes. (Jesus et al., 2019)
MeRIP Seq häufig gestellte Fragen (FAQs)
Könnte MeRIP-seq artenspezifischen Einschränkungen unterliegen?
Für Arten mit Referenzgenomen, die auf chromosomaler Ebene assembliert sind, und solchen mit umfassenden gtf-Annotationsdateien ist diese Methodik anwendbar. Es gibt unterschiedliche Methoden und Parameter, die in der Peak-Erkennungssoftware für Eukaryoten, Prokaryoten oder Viren verwendet werden. Daher ist es ratsam, dass Projekte, die sich auf Prokaryoten oder Viren konzentrieren, vorläufige Bewertungen durchführen.
2. Warum ist es notwendig, Input- und IP-Proben gleichzeitig in MeRIP-Experimenten zu messen?
Das MeRIP-Projekt zeichnet sich durch zwei Komponenten aus: das Input und das IP. Im experimentellen Verfahren nutzt die IP-Probe einen m6A-Antikörper, um gezielt die methylierten RNA-Fragmente anzureichern. Im Gegensatz dazu dient das Input, das ausschließlich aus fragmentierter RNA besteht, als Kontrolle, um Hintergrundgeräusche zu minimieren. Sowohl die IP- als auch die Input-Komponenten werden parallel für den Bibliotheksaufbau und die Sequenzierung verarbeitet. Für die Analyse der Peak-Erkennung müssen die Daten beider Proben integriert werden, wobei die Input-Daten verwendet werden, um Peaks auszuschließen, die mit hohen Basis-Expressionsniveaus oder unspezifischen Bindungsereignissen korrelieren, um die Präzision der Peak-Bestimmung zu maximieren.
3. Was sind die Stärken und Einschränkungen der MeRIP-Sequenzierung?
Die MeRIP-Sequenzierungstechnik bietet mehrere Vorteile:
Hochdurchsatz: Es ermöglicht die gleichzeitige Analyse einer beträchtlichen Anzahl von RNA-Proben. Hohe Sensitivität: Es ist in der Lage, m6A-Modifikationen mit geringer Häufigkeit nachzuweisen. Räumliche Auflösung: Diese Technik kann den Standort von m6A-Modifikationen genau bestimmen.
Die MeRIP-Sequenzierungstechnik hat jedoch auch ihre Einschränkungen:
Antikörperabhängig: Die Auswahl und Qualität der Antikörper können potenziell die experimentellen Ergebnisse beeinflussen.
Präzision: Während die Sequenzierungsdaten einen allgemeinen Standort von m6A-Modifikationen liefern können, sind sie nicht in der Lage, die genaue Base zu bestimmen.
Komplexe Datenanalyse: Ein hohes Maß an spezialisierten Fähigkeiten in der Datenanalyse ist erforderlich, um Sequenzierungsdaten zu interpretieren und m6A-Modifikationsstellen zu identifizieren.
4. Was ist das Prinzip der MeRIP-Sequenzierung?
Das Prinzip der Merip-Sequenzierung basiert auf der MeRIP-Technologie und hochdurchsatzfähiger RNA-Sequenzierung. Zunächst durchlaufen RNA-Proben MeRIP-Experimente, bei denen spezifische Antikörper eingesetzt werden, um RNA-Moleküle zu binden, die mit m6A modifiziert sind. Anschließend werden m6A-modifizierte RNA-Moleküle aus der Gesamt-RNA durch Immunpräzipitation angereichert. Nach der Anreicherung wird die RNA einer reversen Transkription und der Bibliotheksvorbereitung für die DNA-Sequenzierung unterzogen, wobei hochdurchsatzfähige Sequenzierungstechnologie verwendet wird. Schließlich erfolgt die Analyse der Sequenzierungsdaten, um die Positionen und relativen Häufigkeiten der m6A-Modifikationen zu bestimmen.
MeRIP Seq Fallstudien
RNA-Methylome zeigen die m6A-vermittelte Regulation des DNA-Demethylase-Gens SlDML2 bei der Reifung von Tomatenfrüchten.
Journal: Genom Biologie
Impactfaktor: 14,028
Veröffentlicht: 06. August 2019
Hintergründe
Es ist gut etabliert, dass die Modifikationen von DNA 5mC und mRNA m6A eine entscheidende Rolle bei der Regulation der Genexpression spielen. Die Erhaltung der DNA 5mC-Modifikation unterstreicht ihre grundlegende Rolle bei der Modulation mehrerer biologischer Prozesse. Diese Studie hebt ihre Verbindung zur Reifung von Früchten hervor. Frühere Untersuchungen haben gezeigt, dass die Mutagenese des DNA-Demethylase-Gens SlDML2 in Tomaten eine Hypermethylierung im gesamten Genom auslöst und die Fruchtreifung erheblich hemmt. Die Rolle, die m6A in diesem Prozess spielt, sowie die potenziellen Wechselwirkungen zwischen 5mC und m6A bleiben jedoch unklar. Es ist bereits bekannt, dass die m6A-Modifikation, vermittelt durch die RNA-Demethylase SlALKBH2, das homologe Gen der DNA-Demethylase SlDML2 in Arabidopsis regulieren kann.
Methoden
- Extrahieren gesamte RNA
- Bereichern mRNA
- Fragment
- Stellen Sie MeRIP-seq-Bibliotheken her
- Illumina HiSeq X, PE150
- Peak-Erkennung
- Differenzielle Methylierungsanalyse
- Anreicherungsanalyse von Genen, die mit Peaks assoziiert sind
Ergebnisse
1. Die m6A-Modifikation zeigt während des Reifungsprozesses der Früchte eine dynamische Verschiebung, wobei der Gesamtm6A-Spiegel allmählich abnimmt, während die Früchte reifen, was den Mustern der DNA-Methylierung entspricht. Bei Mutationen mit Reifungsdefekten wie Cnr ist der Gesamtm6A-Spiegel neben der DNA-Hypermethylierung ebenfalls höher.
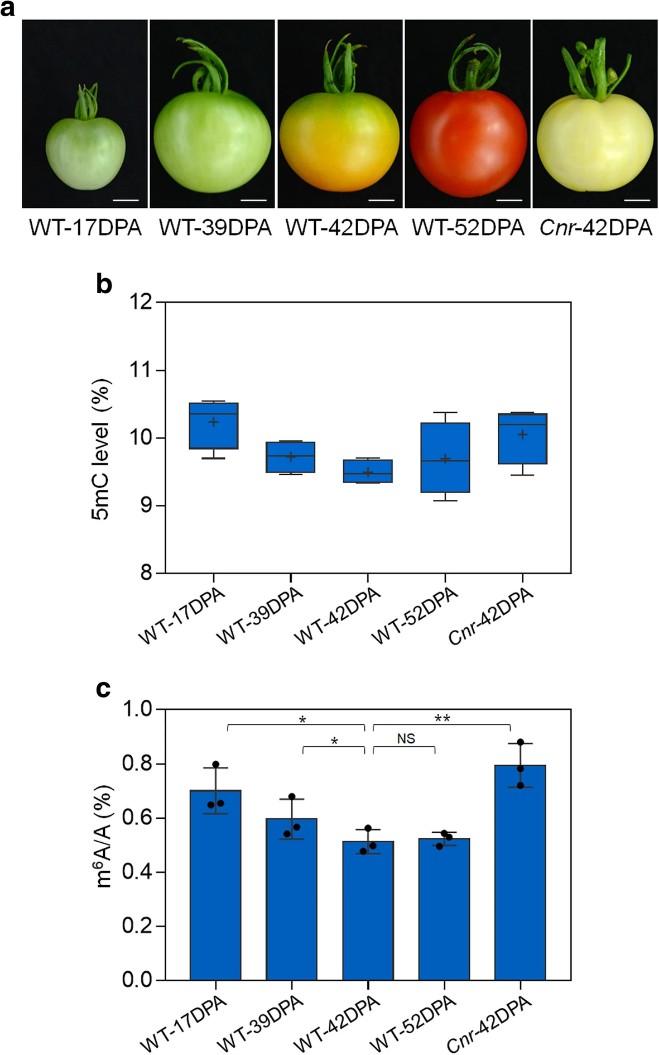 Abbildung 1 Dynamik der DNA-Methylierung (5mC) und der mRNA-m6A-Methylierung bei der Reifung von Tomatenfrüchten.
Abbildung 1 Dynamik der DNA-Methylierung (5mC) und der mRNA-m6A-Methylierung bei der Reifung von Tomatenfrüchten.
2. Die Methylierungsmodifikation m6A ist weit verbreitet in der mRNA von Tomatenfrüchten und zeigt eine umgekehrte Beziehung zu den Transkriptionsniveaus der Gene.
Die Gesamtm6A-Spiegel während der Fruchtreifung und im Cnr-Mutanten entsprechen der Expression des m6A-Demethylase-Gens SlALKBH2, das der DNA-Methylierungsregulation unterliegt. SlALKBH2, eine m6A-Demethylase, hat die Fähigkeit, an die mRNA des DNA-Demethylase-Gens SlDML2 zu binden, um dessen m'6A-Modifikation und Stabilität zu regulieren. Bei einer Mutation des SlALKBH2-Gens steigen die m6A-Spiegel in SlDML2, was mit einem verringerten mRNA-Gehalt einhergeht und somit die normale Fruchtreifung behindert.
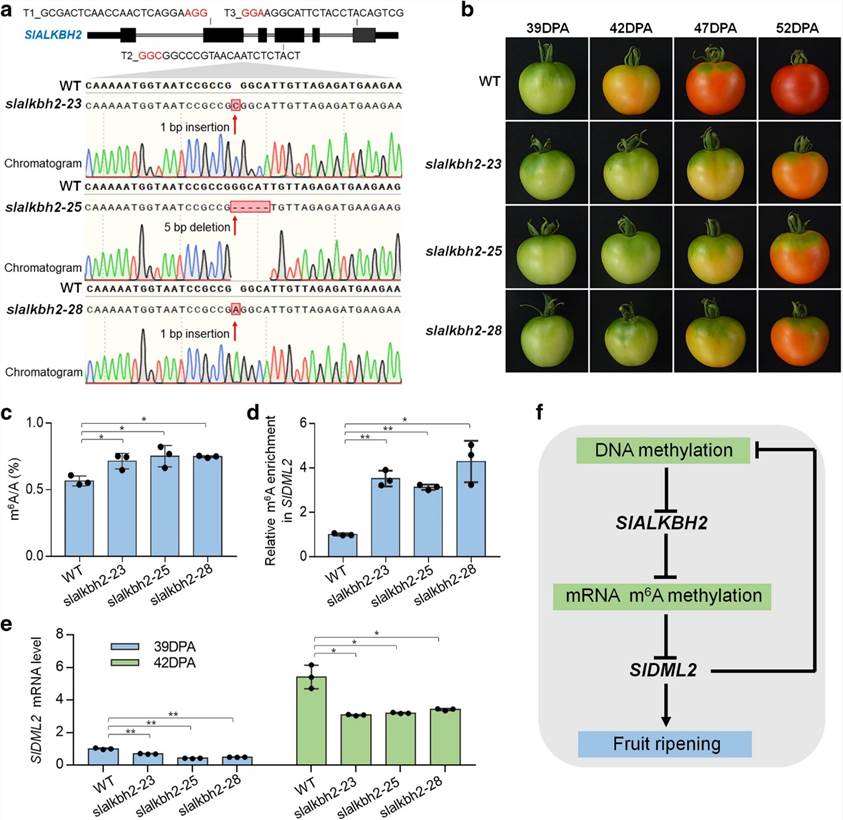 Abbildung 2. SlALKBH2 ist notwendig für die normale Reifung von Tomatenfrüchten.
Abbildung 2. SlALKBH2 ist notwendig für die normale Reifung von Tomatenfrüchten.
Fazit
Diese Studie bestätigt vorläufig die Verbindung zwischen DNA-Methylierung und RNA-Methylierung, indem sie einen neuen Mechanismus der Regulierung des Fruchtreifungsprozesses aufdeckt und neue Einblicke in das Kontrollnetzwerk der Fruchtreife bietet. Angesichts der Multifunktionalität von DNA-Methylierung und RNA-Methylierung wird der in dieser Studie demonstrierte Feedback-Regulierungsmechanismus auch auf andere biologische Prozesse anwendbar sein.
Referenz
- Zhou L, Tian S, Qin G. RNA-Methylome zeigen die m 6 A-vermittelte Regulation des DNA-Demethylase-Gens SlDML2 bei der Reifung von Tomatenfrüchten. Genombiologie, 2019, 20: 1-23.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Die Spaltung von Phagen-DNA durch Restriktionsendonukleasen ermöglicht die Wiederbelebung aus der durch Cas13 induzierten bakteriellen Dormanz.
Zeitschrift: Nature Mikrobiologie
Jahr: 2023
IL-4 fördert die Erschöpfung von CD8.+ CART-Zellen
Zeitschrift: Nature Communications
Jahr: 2024
Fettreiche Diäten während der Schwangerschaft führen zu Veränderungen der DNA-Methylierung und Proteinexpression im Pankreasgewebe der Nachkommen: Ein Multi-Omics-Ansatz
Internationale Zeitschrift für Molekulare Wissenschaften
Jahr: 2024
KMT2A assoziiert mit dem PHF5A-PHF14-HMG20A-RAI1 Subkomplex in Stammzellen des Bauchspeicheldrüsenkrebses und reguliert epigenetisch deren Eigenschaften.
Zeitschrift: Nature Communications
Jahr: 2023
Krebsassoziierte DNA-Hypermethylierung von Polycomb-Zielen erfordert die doppelte Erkennung von Histon H2AK119-Ubiquitinierung und der sauren Tasche des Nukleosoms durch DNMT3A.
Journal: Wissenschaftliche Fortschritte
Jahr: 2024
Genomisches Imprinting-ähnliches monoalleles väterliches Ausdrucksmuster bestimmt das Geschlecht von Kanalwelsen.
Journal: Wissenschaftliche Fortschritte
Jahr: 2022
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


