
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Lange Amplicon-Analyse (LAA)
CD Genomics bietet einen professionellen und kosteneffizienten LAA-Service mit einer Sequenzierungstiefe von <500 bis 10K CCSs pro Probe, um Ihre spezifischen Forschungsanforderungen zu erfüllen.
Die Einführung der Long Amplicon Analyse
Die Long Amplicon Analyse (LAA) ist eine molekularbiologische Technik, die entwickelt wurde, um erweiterte DNA-Sequenzen zu untersuchen und zu analysieren. Diese Strategie amplifiziert hauptsächlich lange Fragmente genomischer DNA durch die Polymerase-Kettenreaktion (PCR), bevor eine anschließende Sequenzierung und Dateninterpretation erfolgt. Diese Methode hat bedeutende Anwendungen in der Genomik, Evolutionsbiologie, Krankheitsforschung sowie in anderen Bereichen, die Forschung zu komplexen Genstrukturen und Variationen erfordern.
Basierend auf den PacBio Circular Consensus Sequences (CCSs) kann die Polymerase dasselbe DNA-Fragment mehrmals kopieren, was hochfidelitätsreiche, lange Reads (>99,9% Genauigkeit der Einzelmolekül-Reads) erzeugt. Die Long Amplicon Analysis (LAA) unter Verwendung von Einzelmolekül, Echtzeit (SMRT) Sequenzierung und das Sequel-System erzeugt hochpräzise und phasierte CCSs aus langen Amplicons.
 Abbildung 1. Übersicht über die SMRT-Sequenzierungstechnologie. (Simon Ardui, u. a.., 2018)
Abbildung 1. Übersicht über die SMRT-Sequenzierungstechnologie. (Simon Ardui, u. a.., 2018)
Im Vergleich zu den Plattformen für Kurzlesesequenzierung ermöglicht die Langlesesequenzierung von PacBio eine einfache Sequenzierung von Ampliconen oder erfassten Fragmenten, die von mehreren hundert Basenpaaren bis zu 10 Kb reichen. Diese langen Sequenzen sind sehr nützlich für die Visualisierung von Varianten, einschließlich SNPs, CNVs und anderen strukturellen Varianten, die typischerweise keine Assemblierung erfordern.
Was sind die Vorteile der Langampliconanalyse?
- Hohe Genauigkeit: Durch den Einsatz von hochfidelitäts Polymerasen reduziert die Long Amplicon Analyse Fehler während des Amplifikationsprozesses und verbessert die Zuverlässigkeit der Ergebnisse.
- Komplexe Regionsauflösung: LAA kann komplexe und sich wiederholende Regionen im Genom abdecken und erläutern, wodurch ein umfassenderes Set an genomischen Informationen bereitgestellt wird.
- Breite Anwendbarkeit: Die Technik ist nicht nur für nukleare Genome anwendbar, sondern auch nützlich für die Analyse verschiedener DNA-Typen, einschließlich mitochondrialer DNA und chloroplastärer DNA.
- Hohe Nachweisempfindlichkeit: Die Analyse langer Amplicons kann große Insertionen/Löschungen, Inversionen und Translokationen identifizieren und nachweisen, wodurch die Empfindlichkeit für die Erkennung komplexer Genomvariationen erhöht wird.
- Hohe haplotypische Auflösung: Langzeit-Sequenzierung kann verschiedene Haplotypen desselben Individuums innerhalb eines einzelnen Amplicons identifizieren und unterscheiden und liefert detaillierte Informationen über die Haplotypstruktur und -variationen.
- Vollständige Genlängenabdeckung: Bei Mutationen, die mehrere Exons oder ganze Gene betreffen, kann LAA gesamte Genregionen abdecken und sicherstellen, dass keine wichtigen Mutationen übersehen werden.
- Vereinfachter Zusammenbauprozess: Die Long-Read-Fähigkeit deckt größere genomische Regionen ab, wodurch Fehlanpassungen und Redundanzen während des Zusammenbauprozesses verringert werden, was die Zusammenbauergebnisse präzise und zuverlässig macht.
Was sind die Anwendungen der Lang-Amplikon-Analyse?
- Vollständige 16S/18S/ITS-Gensequenzierung
- Vollständig HLA-Typisierung
- Alternative Haplotypisierung
- De novo Zusammenstellung des gezielten Gens
- Benutzerdefiniert Amplicon-Sequenzierung
Lang-Amplikon-Analyse-Workflow
Dienstspezifikation
Beispielanforderungen
|
|
 |
Sequenzierung
|
 |
Bioinformatikanalyse
Für die vollständige Sequenzierung der 16S/18S/ITS-Gene:
|
Analyse-Pipeline
Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details zur Lang-Amplikonanalyse für Ihr Schreiben (Anpassung)
Referenzen:
- Simon Ardui, u. a.(2018) Die Einzelmolekül-Echtzeit-Sequenzierung (SMRT) erreicht ihre Reife: Anwendungen und Nutzen für medizinische Diagnosen. Nukleinsäurenforschung46(5) 2159–2168.
- Joshua P. Earl, u. a.(2018) Artenebene Profilierung der bakteriellen Gemeinschaft des gesunden sinonasalen Mikrobioms unter Verwendung der Pacific Biosciences-Sequenzierung von vollständigen 16S rRNA-Genen. Mikrobiom. 6.
- Henk P.J. Buermans, u. a.(2017) Flexible und skalierbare Full-Length CYP2D6 Long Amplicon PacBio-Sequenzierung. Menschliche Mutation38(3) 310–316.
- Frans, Glynis, u. a.(2018) Konventionelle und Einzelmolekül-Target-Sequenzierungsmethode zur spezifischen Variantenerkennung in IKBKG unter Umgehung des IKBKGP1-Pseudogens. Das Journal für Molekulare Diagnostik 20(2): 195-202.
Demonstrationsergebnisse
 (Französisch u. a.., 2018)
(Französisch u. a.., 2018)
Häufig gestellte Fragen zur Analyse langer Amplicons (LAA)
1. Was sind die Hauptanwendungsbereiche der Lang-Amplikon-Analyse?
Die Analyse langer Amplicons findet umfangreiche Anwendung in einer Vielzahl von Forschungsbereichen, zu denen gehören:
- Studien zu genomischen strukturellen Variationen: Es wird verwendet, um komplexe Variationen zu analysieren, die bedeutende Insertionen, Deletionen, Inversionen und wiederholte Sequenzen umfassen.
- Forschung zu genetischen Krankheiten: Dieses Werkzeug unterstützt bei der Identifizierung komplexer Mutationen, die mit verlängerten genomischen Regionen verbunden sind, die für die klinische Diagnose relevant sind.
- Studien zur Evolution und Phylogenie: Die Analyse langer Amplicons ermöglicht die Erforschung genetischer Variationen zwischen Arten und wirft somit Licht auf evolutionäre Beziehungen.
- Forschung zur mikrobiellen Vielfalt: Sie wird verwendet, um die Struktur des gesamten Genoms mikrobieller Gemeinschaften in verschiedenen Umweltproben zu untersuchen.
2. Was ist der grundlegende Arbeitsablauf der Long Amplicon Analyse?
Die Kernprozesse, die der Long Amplicon Analyse zugrunde liegen, beginnen mit der DNA-Extraktion und -Reinigung. Der Vorgang umfasst die Isolierung von DNA höchster Qualität aus den gegebenen Proben. Darauf folgt ein PCR-Amplifikationsprozess, bei dem spezifische Primer in Verbindung mit hochfidelitäts Polymerasen verwendet werden, um Zielsegmente zu verstärken.
Anschließend erfolgt der Prozess der Ampliconreinigung, bei dem verbleibende nicht amplifizierte DNA und Primer-Dimere entfernt werden. Dies führt zum Schritt der Bibliotheksvorbereitung und einer Qualitätskontrolle. Hier werden Bibliotheken vorbereitet, die mit Sequenzierungsplattformen kompatibel sind, und eine strenge Qualitätskontrolle wird durchgeführt, um ihre Eignung für nachfolgende Verfahren sicherzustellen.
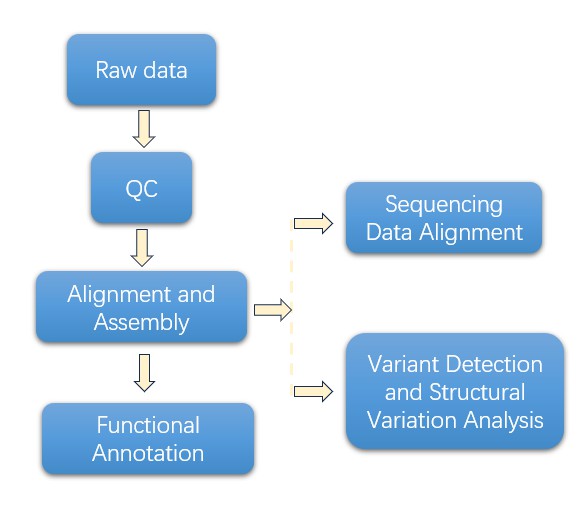
Hochdurchsatz-Sequenzierung ist die nächste Schlüsselphase, die Plattformen wie PacBio nutzt und Oxford Nanopore geschickt in Langzeit-SequenzierungNach dem Sequenzierungsschritt wird eine umfassende Datenanalyse durchgeführt. Diese Analyse umfasst Qualitätskontrolle, Ausrichtung, Variantenentdeckung und funktionale Annotation.
Bevor die Daten gespeichert und für die Verteilung verfügbar gemacht werden, gibt es eine wichtige Phase der Ergebnisvalidierung und -berichterstattung. Wichtige Erkenntnisse werden experimentell verifiziert, und umfassende Analyseberichte werden erstellt. Als abschließender Schritt werden die Daten sicher in Datenbanken gespeichert, wonach sie gemäß den Forschungsanforderungen für die gemeinsame Nutzung zugänglich gemacht werden.
3. Welche häufig verwendeten Werkzeuge gibt es in der Datenanalyse von langen Amplicon-Analysen?
Werkzeuge, die in der Datenanalyse verwendet werden, umfassen:
Qualitätskontrolle: FastQC, Trimmomatic, Cutadapt.
Ausrichtung und Zusammenbau: BWA, Bowtie2, Minimap2, Canu, Flye.
Variantenerkennung: GATK, FreeBayes, Manta, LUMPY.
Funktionale Annotation: ANNOVAR, SnpEff.
4. Wie kann die Genauigkeit und Zuverlässigkeit der Ergebnisse der Long Amplicon Analyse sichergestellt werden?
Mehrere methodologische Protokolle können implementiert werden, um Genauigkeit und Zuverlässigkeit zu stärken, wie zum Beispiel:
- DNA-Extraktion und -Reinigung: Die Verwendung von anspruchsvollen Kits in Verbindung mit einer sorgfältigen Durchführung der Verfahren kann eine hochwertige DNA-Extraktion und -Reinigung ermöglichen.
- Optimierung der PCR-Bedingungen: Die Feinabstimmung spezifischer Bedingungen im Polymerase-Kettenreaktionsprozess (PCR), wie z.B. der Annealing-Temperatur und der Verlängerungszeit, kann dazu beitragen, eine effiziente Segmentamplifikation zu gewährleisten.
- Hochfidelitäts-Polymerase-Nutzung: Die Anwendung von Hochfidelitäts-Polymerasen spielt eine wichtige Rolle bei der Minimierung von Amplifikationsabweichungen.
- Aufmerksame Qualitätskontrolle: Strenge Qualitätskontrollmaßnahmen sollten an kritischen Punkten während der Sequenzierung und der Datenanalysephasen beachtet werden.
- Experimentelle Validierung: Entscheidende Ergebnisse sollten experimentell validiert werden. Dies könnte Methoden wie umfassen Sanger-Sequenzierung oder quantitative PCR (qPCR).
Fallstudien zur Analyse langer Ampliconen (LAA)
Definition von Referenzallelen für Blutgruppengene durch Langzeit-Sequenzierung: Machbarkeitsnachweis in der ACKR1 Gen, das die Duffy-Antigene kodiert
Zeitschrift: Transfusionsmedizin und Hämostaseologie
Impactfaktor: 2,283
Veröffentlicht: 11. Dezember 2019
Hintergrund
Im aufstrebenden Bereich der Blutgruppengenomik stellen die Definition und Neudefinition von Referenzgenen oder Allelsequenzen für verschiedene Blutgruppengene bedeutende Ziele dar, sowohl für diagnostische Anwendungen als auch für wissenschaftliche Erkundungen. Angesichts des Aufkommens innovativer, leistungsstarker Sequenzierungstechnologien haben wir uns bemüht, die Variabilität der drei prominentesten Allele von ACKR1 - das Gen, das klinisch kritische Duffy-Antigene kodiert - auf Haplotyp-Ebene, unter Verwendung eines Long-Read-Sequenzierung Methodologie.
Methoden
- Genomische DNA (gDNA) Proben
- Blutproben
- Genomische DNA-Extraktion
- LR-PCR Amplifikation
- Bibliotheksvorbereitung
- SMRT-Sequenzierung
- Fortsetzungs-System
- Datenqualitätskontrolle
- Long-Amplicon-Analyse (LAA) Modul
- Variant-Analyse
Ergebnisse
Die Autoren erhielten hochwertige Sequenzierungsdaten für die 162 Allele (Genauigkeit >0,999). Insgesamt wurden zweiundzwanzig Nukleotidvariationen identifiziert, die in renommierten Datenbanken verzeichnet sind. Diese Variationen definierten 19 Haplotypen: bestehend aus vier in 46. ACKR1*01acht in 63 ACKR1*02und sieben in 53 ACKR1*02N.01 Allele jeweils.
 Abb. 1. Langstrecken-PCR (LR-PCR) Amplifikation des gesamten ACKR1 Genlocus.
Abb. 1. Langstrecken-PCR (LR-PCR) Amplifikation des gesamten ACKR1 Genlocus.
Tabelle 1. ACKR1 Varianten und Haplotypen, die durch Langzeit-Sequenzierung identifiziert wurden

Fazit
Entsprechend wurde ein Kader spezifischer Referenzallele eingerichtet, der auf der Sequenzierungstechnologie der dritten Generation basiert und eine umfassende longitudinale Untersuchung von Genloci über mehrere tausend Basenpaare hinweg ermöglicht. Diese neuartige Technik ergänzt die Sequenzierungstechniken der zweiten Generation oder Kurzlesetechniken und erweist sich als entscheidend für die Entschlüsselung neuer, seltener und null Allele.
Referenz:
- Fichou, Y., Fichou, Y., Berlivet, I., Richard, G., u. a.. Definition von Referenzallelen für Blutgruppengene durch Langzeit-Sequenzierung: Machbarkeitsnachweis in der ACKR1 Gen, der die Duffy-Antigene kodiert. Transfusionsmedizin und Hämostaseologie, 2020, 47(1): 23-32.
Verwandte Veröffentlichungen
Hier sind einige Veröffentlichungen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Bakterielle Gemeinschaften von Cassiopea in den Florida Keys teilen sich wichtige bakterielle Taxa mit Korallen-Mikrobiomen.
Journal: bioRxiv
Jahr: 2024
Produktion eines Bakteriocin-ähnlichen Proteins PEG 446 aus Clostridium tyrobutyricum NRRL B-67062
Journal: Probiotika und antimikrobielle Proteine
Jahr: 2024
Entwirrung der Rolle von Pathobionten aus Bacteroides-Arten bei entzündlichen Darmerkrankungen
Journal: bioRxiv
Jahr: 2023
Eine Chromosomenebene-Genomressource zur Untersuchung von Virulenzmechanismen und der Evolution des Kaffeerostpathogens Hemileia vastatrix
Journal: bioRxiv
Jahr: 2022
Streptomyces buecherae sp. nov., ein Actinomycet, isoliert aus mehreren Fledermausarten
Journal: Antonie van Leeuwenhoek
Jahr: 2020
Mehr ansehen Artikel, die von unseren Kunden veröffentlicht wurden.


