
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
CRISPR-Screen-Sequenzierung
Einführung in die CRISPR-Screen-Sequenzierung
Effektives CRISPR-Screening beginnt mit einer sorgfältig gestalteten Bibliothek von single guide RNAs (sgRNAs), die auf spezifische Gene oder Loci abzielen. Der Designprozess umfasst die Auswahl geeigneter Sequenzen, die Synthese der sgRNAs und anschließend das Klonen in Vektoren oder das Transkribieren in RNA für die Zelltransfektion. Das Screening erfolgt dann mithilfe von positiven oder negativen Selektionsmethoden. Nach der Transfektion werden die Zellen physisch in verschiedene Populationen sortiert, gefolgt von PCR-Amplifikation und hochdurchsatzfähiger Parallelsequenzierung. Die Datenanalyse identifiziert anschließend die sgRNAs und die entsprechenden Zielgene von Interesse.
Die Nutzung von CRISPR-Screening-Technologie für Hochdurchsatz-Assays ermöglicht die Erstellung umfangreicher mutantischer Zellbibliotheken, die verschiedenen externen Bedingungen ausgesetzt werden können, um mutantische Zelluntergruppen zu isolieren. Hochdurchsatz-Sequenzierung und bioinformatische Analysen verdeutlichen weiter die Beziehungen zwischen Phänotypen und Genotypen. Dieses auf CRISPR/Cas9 basierende Hochdurchsatz-Screening bietet erhebliche Vorteile gegenüber RNA-Interferenz (RNAi)-Screening-Techniken, einschließlich der Überwindung von Problemen im Zusammenhang mit niedriger Transfektionseffizienz und der begrenzten Fähigkeit, die Genexpression auf mRNA-Ebene zu unterdrücken.
In der Ära der Präzisionsmedizin hat das CRISPR-Screening einen enormen wissenschaftlichen Wert und bietet großes Potenzial für Forschungsanwendungen. Diese Methode bietet einen robusten, einfachen und programmierbaren Ansatz für die großflächige Genbearbeitung – oft als genomweite oder hochdurchsatzfähige Genbearbeitung bezeichnet – die eine erhebliche Wirksamkeit gezeigt hat, insbesondere in der Forschung an Säugetieren.
Um den aufkommenden Bedürfnissen der Forschungsgemeinschaften gerecht zu werden, hat CD Genomics eine kostengünstige, zuverlässige und hochdurchsatzfähige Strategie für die CRISPR-Screen-Sequenzierung entwickelt, indem ampliconbasierte Next-Generation-Sequenzierung genutzt wird. Unser CRISPR-Screen-Sequenzierungsdienst kann Ihnen direkte und detaillierte Informationen über sgRNA und die Analyse gezielter Gene sowie funktionale Anreicherungsanalysen bieten, um Forschern zu helfen, die Funktionen von Genen auf hochdurchsatzfähige Weise zu untersuchen.
 Abbildung 1. Übersicht über CRISPR-Screening.
Abbildung 1. Übersicht über CRISPR-Screening.
Vorteile unseres CRISPR-Screen-Sequenzierungsdienstes
- Effizientes Screening: Es bearbeitet mehrere Gene gleichzeitig, was ein effektives Screening und die Bewertung von Genfunktionen ermöglicht.
- Umfassende Bewertung: Bietet einen systematischen Ansatz zur Bewertung von Genfunktionen innerhalb von Zellen oder Organismen.
- Präzise genetische Modifikationen: Zielt auf spezifische genomische Bereiche ab, um genaue Genbearbeitungen wie Knockouts oder Mutationen zu erreichen.
- Schnelle Dateninterpretation: Nutzt fortschrittliche Sequenzierungstechnologie für eine zügige und präzise Datenanalyse.
- Vielseitige Anwendungen: Weit verbreitet bei der Entdeckung von Arzneimittelzielen, der Etablierung von Krankheitsmodellen und der Förderung der therapeutischen Entwicklung.
- Optimierte Prozesse: Steigert die Effizienz und senkt die Kosten im Vergleich zu herkömmlichen Methoden, wodurch Forschungsprojekte beschleunigt werden.
- Umfangreiche Multiplexing-Flexibilität und Hochdurchsatz-Sequenzierung ermöglichen die Quantifizierung und den Vergleich der Frequenzen von sgRNAs.
- Kosteneffektiv und hochsensible Nachweisniveaus ohne Verzerrung.
- Keine mühsamen und zeitaufwändigen Klonierungsschritte erforderlich.
- Engagierte Unterstützung von spezialisierten Wissenschaftlern auf Doktoratsniveau.
Anwendungen der CRISPR-Screen-Sequenzierung
- Genfunktionen Studien: Die CRISPR-Screen-Sequenzierung ermöglicht es Forschern, systematisch die Funktionen von Genen in Zellen oder Organismen zu untersuchen, insbesondere deren Rollen in komplexen biologischen Prozessen und der Krankheitsentwicklung.
- Arzneimittelzielentdeckung: Durch großangelegte Screening- und Analyseverfahren von genomischen Editierungen können potenzielle Arzneimittelziele entdeckt und hinsichtlich ihrer Rolle in der Krankheitsbehandlung bewertet werden.
- Etablierung von Krankheitsmodellen: Basierend auf den Ergebnissen des CRISPR-Screen-Sequenzierens können genauere Krankheitsmodelle zur Untersuchung von Krankheitsmechanismen und zur Screening von therapeutischen Ansätzen etabliert werden.
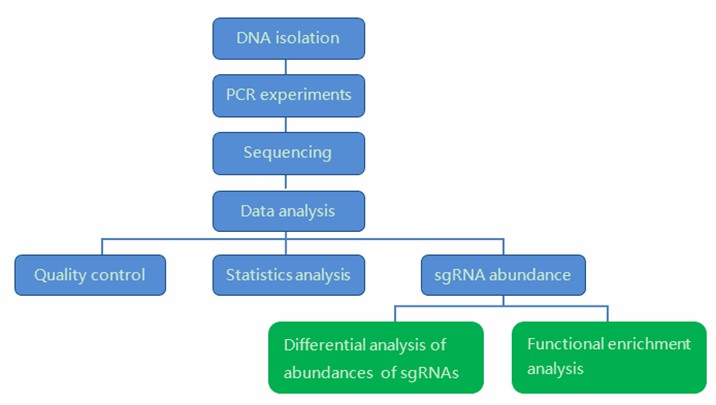
CRISPR-Screen-Sequenzierungs-Workflow
Unser hochqualifiziertes Expertenteam führt das Qualitätsmanagement nach jedem Verfahren durch, um umfassende und genaue Ergebnisse sicherzustellen. Unser CRISPR-Screen-Sequenzierungsworkflow ist unten aufgeführt, einschließlich DNA-Isolierung, PCR-Experimenten, Sequenzierung und bioinformatischer Analyse. Unsere CRISPR-Screen-Sequenzierung kann Forschern auch helfen, herauszufinden, welche Gen-Knockouts Phänotypen hervorgebracht haben, die für den Screen relevant sind.

Dienstspezifikationen
Beispielanforderungen
|
|
 Klicken |
Sequenzierungsstrategie
|
 |
Bioinformatikanalyse
Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an:
|
Analyse-Pipeline
Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details in CRISPR-Screen-Sequenzierung für Ihre Schreibanpassung
CD Genomics bietet Dienstleistungen zur Identifizierung von sgRNA-Sequenzen und Zielgenen von Interesse an, gefolgt von einer Tiefensequenzierung zu äußerst wettbewerbsfähigen Preisen. Unsere Fähigkeiten ermöglichen die gleichzeitige Sequenzierung von Hunderten von Proben. Wir nutzen das MAGeCK-Tool zur Analyse der sgRNA-Häufigkeit und der differentiellen Expression zwischen Gruppen. Sollten Sie weitere Anforderungen oder Anfragen haben, zögern Sie bitte nicht, uns zu kontaktieren.
Referenzen
- Ella H, u. a.Genetische Screens und funktionelle Genomik mit CRISPR-Cas9-Technologie. FEBS Journal, 2015, 1383-1393.
- Zhou Y, u. a.Hochdurchsatz-Screening einer CRISPR/Cas9-Bibliothek für funktionelle Genomik in menschlichen Zellen. Natur, 2014, 509(2):487-491.
Demonstrationsergebnisse
Teilweise Ergebnisse sind unten aufgeführt:
CRISPR-Screen-Sequenz FAQs
1. Wie entwirft man sgRNA-Bibliotheken für Ganzgenom- oder Teilgenomsequenzen?
Das Design von sgRNA-Bibliotheken für die Ganzgenom- oder Teilgenomsequenzierung ist ein entscheidender Aspekt für die effektive Nutzung der CRISPR-SCREEN-Technologie. Um eine hohe Editierungseffizienz, minimierte Off-Target-Effekte und das Sicherstellen von Frameshift-Mutationen bei der Gen-Knockout zu erreichen, ist eine umfassende Bewertung mehrerer Faktoren erforderlich. Die folgenden Schlüsselaspekte sind für ein optimales sgRNA-Design unerlässlich:
- Analyse des GC-Gehalts;
- Auswahl der Schnittpositionen;
- Analyse der Sekundärstrukturen in der Sequenz;
- Bewertung der gRNA-Spezifität;
- Vermeidung von Sequenzen im gRNA (außer PAM) mit vier oder mehr aufeinanderfolgenden T-Nukleotiden in der Zielsequenz;
- Vorhersage des Effizienzscores der gRNA-Sequenz;
- Bewertung potenzieller Off-Target-Effekte und Fehlanpassungen in der gRNA-Sequenz.
2. Welche Arten von CRISPR-Screenings können durchgeführt werden?
- Positive SelektionsscreeningsIdentifizieren Sie Gene, deren Störung einen Wachstumsvorteil oder Widerstand unter bestimmten Bedingungen verleiht.
- Negative AuswahlbildschirmeIdentifizieren Sie Gene, deren Störung zu einem Verlust der Lebensfähigkeit oder Empfindlichkeit gegenüber bestimmten Behandlungen führt.
- Gemeinsame BildschirmeNutzen Sie eine gemischte Zellpopulation, die mit verschiedenen sgRNAs transduziert wurde, um eine Hochdurchsatzanalyse zu ermöglichen.
- Angeordnete BildschirmeJede Vertiefung oder Probe enthält Zellen, die mit einem einzelnen sgRNA transduziert wurden, was eine detaillierte phänotypische Analyse ermöglicht.
3. Wie stellen Sie minimale Off-Target-Effekte bei der CRISPR-Screen-Sequenzierung sicher?
- sgRNA-DesignVerwenden Sie bioinformatische Werkzeuge, um sgRNAs mit hoher Spezifität für Zielstellen und minimalen vorhergesagten Off-Target-Stellen zu entwerfen.
- ValidierungFühren Sie Experimente durch, um die Spezifität der ausgewählten sgRNAs zu validieren.
- Hochpräzise Cas9-VariantenNutzen Sie Cas9-Varianten, die für reduzierte Off-Target-Aktivität entwickelt wurden.
4. Wie analysieren Sie die Daten aus der CRISPR-Screen-Sequenzierung?
Die Datenanalyse umfasst mehrere Schritte:
- Qualitätskontrolle von SequenzierungsdatenÜberprüfen Sie die Qualität der Sequenzierungsreads.
- Mapping-LesungenRichten Sie die Reads an das Referenzgenom aus, um sgRNA-Sequenzen zu identifizieren.
- TrefferidentifikationBestimmen Sie, welche sgRNAs im Vergleich zu den Kontrollen signifikant angereichert oder vermindert sind.
- FunktionalanalysisInterpretieren Sie die biologische Bedeutung der identifizierten Treffer, häufig unter Verwendung von Bioinformatik-Tools zur Analyse von Genwegen und Interaktionen.
CRISPR-Screen-Sequenz-Fallstudien
Genomweite CRISPR/Cas9-Bibliotheksuntersuchung identifiziert PCMT1 als einen entscheidenden Treiber der Metastasierung von Eierstockkrebs.
Zeitschrift: Journal für experimentelle und klinische Krebsforschung
Impactfaktor: 12,568
Veröffentlicht: 15. Januar 2022
Hintergrund
Die Metastasierung von Krebs umfasst die Migration und Invasion von Tumorzellen, was eine Resistenz gegen Anoikis erfordert. Die extrazelluläre Matrix (ECM) spielt dabei eine entscheidende Rolle. Durch CRISPR/Cas9-Knockout-Screenings in Ovarialkarzinomzellen wurde PCMT1 als entscheidend für die Anoikis-Resistenz identifiziert. PCMT1 interagiert mit dem ECM-Protein LAMB3 und aktiviert Signalwege, die bei der Zelladhäsion und Invasion helfen. Ein erhöhtes PCMT1 in Metastasen hebt sein Potenzial als therapeutisches Ziel hervor.
Materialien & Methoden
Probenvorbereitung
- Patienten
- Gewebeproben
- RNA-Extraktion
Sequenzierung
- Zellkultur
- Genomweite CRISPR/Cas9-Screen
- Next-Generation-Sequenzierung (NGS)
- IP-MS
- Quantitative Echtzeit-PCR
- RNAi-Genanreicherungs-Rangfolge (RIGER) Analyse
- Western-Blot-Analyse
- Immunhistochemie (IHC) Analyse
- Statistische Analysen
Ergebnisse
Ein genomweites CRISPR/Cas9-Screening in Ovarialkarzinomzellen identifizierte Gene, die mit Anoikis-Resistenz verbunden sind. Mit der GeCKO v2-Bibliothek wurden SKOV3-Zellen unter Standard- und Ultratiefen-Anhaftungsbedingungen gescreent. Die Sequenzierung ergab 286 Gene aus der negativen Selektion und 122 aus der positiven Selektion. Wichtige Signalwege umfassten die Proteinreparatur und die translationale Initiation, wobei auch bekannte Gene der Anoikis-Resistenz wie RAN, KIF11 und ECT2 identifiziert wurden.
 Abb. 1. Ein zusammengeführter genomweiter CRISPR-Screen in einem Metastasenmodell von Eierstockkrebszellen.
Abb. 1. Ein zusammengeführter genomweiter CRISPR-Screen in einem Metastasenmodell von Eierstockkrebszellen.
IP-MS identifizierte, dass PCMT1 mit LAMB3 interagiert. Unter den 39 hochkonfidenten PCMT1-interagierenden Proteinen war LAMB3 mit Eigenschaften der Zellmetastase verbunden. Funktionale Studien bestätigten die Interaktion zwischen PCMT1 und LAMB3, und das Herunterregulieren von LAMB3 reduzierte die Bildung von Zell-Sphäroiden und die Migration in PCMT1-überexprimierenden Ovarialkarzinomzellen, was darauf hindeutet, dass LAMB3 ein nachgeschaltetes Ziel von PCMT1 bei der Krebsmetastase ist.
 Abb. 2. IP-MS zeigt, dass PCMT1 mit LAMB3 interagiert.
Abb. 2. IP-MS zeigt, dass PCMT1 mit LAMB3 interagiert.
Fazit
Zusammenfassend identifiziert diese Studie PCMT1 als einen entscheidenden Treiber der Anoikis-Resistenz, der die Zelladhäsion, Migration und Invasion verbessert. PCMT1 reguliert die FAK-Src-Signalgebung hoch, fördert die Metastasierung und korreliert mit dem metastatischen Stadium bei Eierstockkrebs. Erhöhte Serum-PCMT1-Spiegel könnten auf ein metastatisches Potenzial hinweisen, und die gezielte Behandlung von PCMT1 mit Antikörpern zeigt vielversprechende Ansätze als therapeutische Strategie gegen die Metastasierung von Eierstockkrebs.
Referenz
- Zhang J, Li Y, Liu H, et al. Genomweite CRISPR/Cas9-Bibliotheksuntersuchung identifiziert PCMT1 als einen entscheidenden Treiber der Metastasenbildung bei Eierstockkrebs. Journal für Experimentelle & Klinische Krebsforschung, 2022, 41(1): 24.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Die HLA-Klasse-I-Immunopeptidome der AAV-Kapsidproteine
Zeitschrift: Frontiers in Immunologie
Jahr: 2023
Isolation und Charakterisierung neuer menschlicher Trägerpeptide aus zwei wichtigen Impfstoff-Immunogenen
Journal: Impfstoff
Jahr: 2020
Änderung des Gewichts, des BMI und der Körperzusammensetzung in einer bevölkerungsbasierten Intervention im Vergleich zu einer genbasierten Intervention: Die NOW-Studie
Journal: Fettleibigkeit
Jahr: 2020
Sarecyclin hemmt die Proteintranslation im Cutibacterium acnes 70S-Ribosom durch einen Zwei-Stellen-Mechanismus.
Zeitschrift: Nucleic Acids Research
Jahr: 2023
Identifizierung eines Darmkommensalen, der die blutdrucksenkende Wirkung von Ester-Angiotensin-Converting-Enzym-Hemmern beeinträchtigt.
Zeitschrift: Hypertonie
Jahr: 2022
Eine Splice-Variante im SLC16A8-Gen führt zu einem Defizit beim Laktattransport in aus menschlichen iPS-Zellen abgeleiteten retinalen Pigmentepithelzellen.
Journal: Zellen
Jahr: 2021
Mehr ansehen Artikel, die von unseren Kunden veröffentlicht wurden.


