
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Tier-/Pflanzen-Exom-Sequenzierungsdienst
Was ist die Exom-Sequenzierung von Tieren/Pflanzen?
Exon-Capture-Sequenzierung ist ein Verfahren, das verwendet wird, um Exons (Sammlungen aller Exons) im Genom zu extrahieren und zu sequenzieren und die Exonvariationen in einer einzelnen Organismensprobe zu erhalten. Dieser Ansatz ermöglicht es der Forschung, sich schnell auf die Teile des Genoms zu konzentrieren, die wahrscheinlich den phänotypischen Variationen Einfluss verleihen. Im Vergleich zur Ganzgenomsequenzierung vereinfacht die Methode der Exon-Capture-Sequenzierung das Genom der Zielarten, reduziert redundante Sequenzen erheblich, kann Kandidatengene, die mit spezifischen Merkmalen in Zusammenhang stehen, genauer und schneller lokalisieren, senkt effektiv die Sequenzierungskosten und wird daher in der Tier- und Pflanzenforschung weit verbreitet eingesetzt.
Die Exom-Sequenzierung von Tieren und Pflanzen spielt eine entscheidende Rolle in verschiedenen Bereichen, einschließlich Genetik, Krankheitsdiagnostik, Evolutionsbiologie und landwirtschaftlicher Züchtung. Durch die gezielte Analyse von protein-kodierenden Regionen ermöglicht dieser Ansatz Forschern, genetische Variationen zu identifizieren, die mit wichtigen Merkmalen assoziiert sind, und liefert Einblicke in erbliche Krankheiten, adaptive Evolution und Artenvielfalt. In der Landwirtschaft beschleunigt die Exom-Sequenzierung die genomische Selektion und unterstützt die Züchtung von Pflanzen und Tieren mit wünschenswerten Eigenschaften wie Krankheitsresistenz und hoher Ertrag. Darüber hinaus fördert sie Naturschutzbemühungen, indem sie die genetische Vielfalt und Resilienz bedrohter Arten analysiert.
Tier- und Pflanzen-Whole-Exom-Serienprodukte
Basierend auf der Hybridisierungsfang-Sequenzierungstechnologie und der Nukleotidsyntheseplattform kann die gezielte Sequenzierung von Exonen von Tieren und Pflanzen weitreichend in der molekularen Züchtung, der Populationsgenetik, der feinen Kartierung von BSA, der Sequenzierung von Mutantenbibliotheken usw. eingesetzt werden.
| Produktname | Referenzgenom | Zielregion Größe |
|---|---|---|
| Weizen Whole-Exom-Erfassungsprodukt | Weichweizen (IWGSC V2.1) | 132,6 MB |
| Gersten-Ganzexom-Erfassungsprodukt | Gerste (MorexV3) | 42,0 MB |
| Masson-Kiefer Ganzexom-Erfassungsprodukt | Kiefer (Pinus tabuliformis) | 111,1 MB |
| Mais Whole-Exome Capture Produkt | Zea mays (B73 V5) | 45,5 MB |
| Maus Whole-Exome Capture Produkt | Hausmaus (mm39) | 38,4 MB |
| Ratten Whole-Exome Capture Produkt | Norwegische Ratte (GRCr8) | 38,3 MB |
| Hundespezifisches Whole-Exome-Capture-Produkt | Hauswolf (canFam4) | 36,0 MB |
| Schweine-Ganzexom-Erfassungsprodukt | Wildschwein (Sscrofa11.1) | 35,8 MB |
| Rinder-Whole-Exom-Capture-Produkt | Hausrind (ARS-UCD2.0) | 36,9 MB |
| Hühnchen Whole-Exom-Capture-Produkt | Hahn (GRCg7b) | 32,1 MB |
CD Genomics bietet umfassende Exom-Sequenzierungsdienste für eine Vielzahl von Tier- und Pflanzenarten an. Durch den Einsatz fortschrittlicher Exon-Erfassungstechnologie und Plattformen der Next-Generation-Sequenzierung (NGS) gewährleisten wir hochwertige Daten zur Identifizierung wichtiger genetischer Variationen.
Vorteile unseres Exom-Sequenzierungsdienstes für Tiere/Pflanzen
Hohe Spezifität: Zielgerichtete Sequenzierung von exonen Regionen oder spezifischen SNP-Stellen von Interesse.
Verbesserte Effizienz: Nutzt Next-Generation-Sequencing (NGS)-Plattformen für schnellere und effizientere Sequenzierung.
Hohe Genauigkeit: Die tiefe Sequenzierungsabdeckung gewährleistet präzise und zuverlässige Ergebnisse.
Kosteneffektiv: Konzentriert sich nur auf Zielregionen und reduziert die Forschungskosten erheblich.
Anwendungen der Exom-Sequenzierung bei Tieren/Pflanzen
Anwendungen der Exom-Sequenzierung bei Pflanzen:
Pflanzenverbesserung: Die Exomfangtechnologie wurde verwendet, um Genvariationen zu identifizieren, die mit Ertrag, Krankheitsresistenz und Anpassungsfähigkeit bei Pflanzen wie Sojabohnen, Reis und Weizen in Zusammenhang stehen. Sie hilft auch beim Erstellen genetischer Karten und QTL-Kartierung.
Mikroexon-Forschung: Mikroexons in Pflanzen spielen eine bedeutende Rolle bei der Regulierung der Genexpression, der Entwicklung und den Krankheitsprozessen.
Komplexe Genomanalyse: Bei komplexen Genomen von Pflanzen reduziert die Exomfangerfassung erheblich die Sequenzierungskosten und verbessert die Datenqualität, insbesondere in Abwesenheit eines Referenzgenoms.
Anwendungen der Exom-Sequenzierung bei Tieren:
Krankheitsforschung und genetische Forschung: Exom-Sequenzierung wird häufig in der Tierkrankheitsforschung und in genetischen Studien eingesetzt, wie zum Beispiel zur Identifizierung von Genvariationen, die mit Phäochromozytomen bei Hunden assoziiert sind, sowie pathogenen Mutationen bei seltenen menschlichen Krankheiten.
Zucht- und Evolutionsstudien: Exom-Sequenzierung hilft, Genvariationen zu identifizieren, die mit produktiven Eigenschaften in Verbindung stehen, und beschleunigt die genetische Verbesserung in der Tierzucht.
Mikroexon-Forschung: Mikroexons bei Tieren sind an der Regulierung der Genexpression und verschiedenen biologischen Prozessen beteiligt.
Tier-/Pflanzen-Exom-Sequenzierungs-Workflow
Die Exom-Sequenzierung umfasst das Extrahieren und Vorverarbeiten von DNA, das Erfassen von Ziel-exonischen Regionen mit spezifischen Sonden, das Anreichern und Amplifizieren der erfassten Fragmente, das Sequenzieren dieser mit Plattformen wie Illumina und das Analysieren der Daten durch das Ausrichten an ein Referenzgenom, um Varianten zu identifizieren, die die Proteinfunktion beeinflussen könnten.

Dienstspezifikationen
| Musteranforderungen
Beispielanforderungen:
|
|

|
Sequenzierung
|
 |
Bioinformatikanalyse
Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an:
|
Analyse-Pipeline
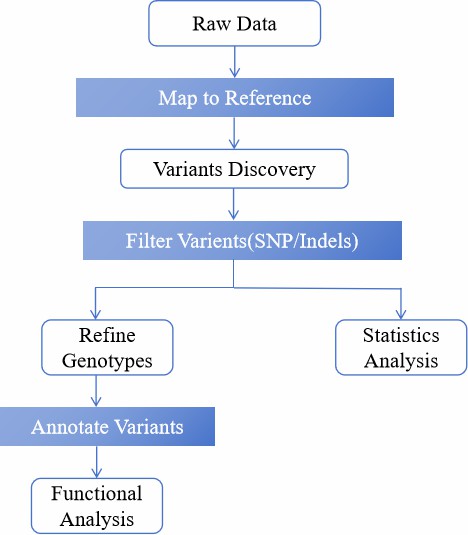
Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details zur gesamten Exom-Sequenzierung für Ihre Schreibanpassung.
CD Genomics bietet umfassende Exom-Sequenzierungsdienste für sowohl Tier- als auch Pflanzenarten an und bietet eine vollständige Lösung von der Probenvorbereitung bis zur fortgeschrittenen bioinformatischen Analyse. Unser Service umfasst die Erfassung exoner Regionen, hochgradige Sequenzierung und präzise Variantenidentifizierung, die auf Ihre spezifischen Forschungsbedürfnisse zugeschnitten sind. Wir nutzen modernste Exon-Erfassungstechnologie und Plattformen der nächsten Generation für die Sequenzierung, um qualitativ hochwertige Ergebnisse sicherzustellen. Egal, ob Sie die Verbesserung von Nutzpflanzen, Krankheitsdiagnosen oder genetische Vielfalt untersuchen, unsere Exom-Sequenzierungsdienste für Tiere und Pflanzen liefern präzise, kosteneffektive Einblicke. Für weitere Informationen oder zur Besprechung individueller Anforderungen können Sie uns gerne kontaktieren.
Demonstrationsergebnisse
Teilweise Ergebnisse sind unten aufgeführt:
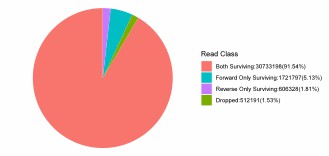
Rohdatenfilterergebnisse.
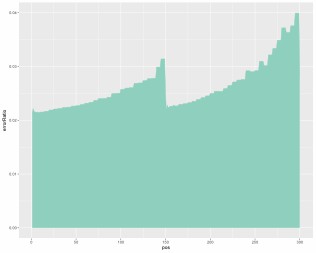
Verteilung der Sequenzierungsfehlerquote.
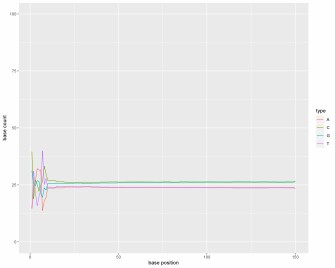
GC-Gehaltverteilung der Proben.
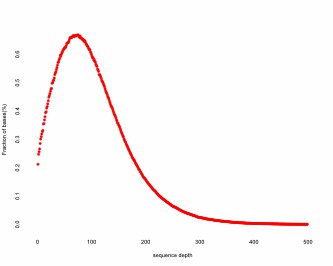
Sequenzierungstiefe.
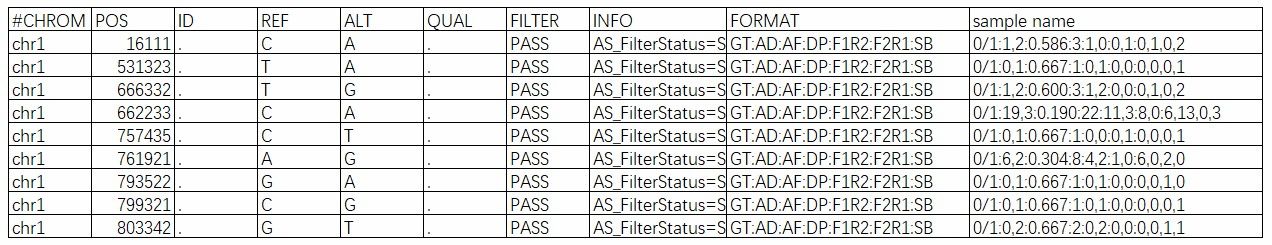
SNV-Identifikationsergebnis.
Tier-/Pflanzen-Exom-Sequenzierungs-FAQs
1.Wie wählt man die geeignete Plattform für das Exom-Sequenzieren von Pflanzen und Tieren aus?
Bei der Auswahl einer Sequenzierungsplattform sollten die folgenden Faktoren berücksichtigt werden:
Zielregionsgröße: Zum Beispiel enthält das Exom von Weizen etwa 170-340 Mb, während das menschliche Exom etwa 30 Mb beträgt.
Probe-Design: Array-basierte Erfassung eignet sich für großangelegte Studien, während die Flüssigphasen-Erfassung besser für kleine Probenmengen oder komplexe Genome geeignet ist.
Sequenzierungstiefe: Die erforderliche Abdeckungstiefe sollte basierend auf den Forschungszielen bestimmt werden. Zum Beispiel wird in der Regel eine höhere Abdeckungstiefe für die Krankheitsdiagnose benötigt.
2. Was sind die zukünftigen Entwicklungstrends der Exomsequenzierung von Pflanzen und Tieren?
Mit den Fortschritten in der Sequenzierungstechnologie wird die Exomsequenzierung von Pflanzen und Tieren in Zukunft effizienter und kostengünstiger werden. Zum Beispiel:
Multispezies-Vergleichsanalyse: Durch die Integration von Exomdaten verschiedener Arten können umfassendere evolutionäre und funktionale Beziehungen aufgedeckt werden.
KI-gestützte Analyse: Nutzung von maschinellen Lernalgorithmen zur Verbesserung der Effizienz und Genauigkeit der Datenanalyse.
3. Was ist der Vorteil der Exom-Sequenzierung gegenüber anderen genomischen Methoden?
Die Hauptvorteile der Exom-Sequenzierung sind die geringeren Kosten im Vergleich zur gesamten Genomsequenzierung und der Fokus auf Exons, die die funktional bedeutendsten Regionen des Genoms sind. Dies macht sie besonders nützlich zur Identifizierung von Mutationen, die die Genfunktion beeinflussen.
4.Wie analysieren Sie die Daten aus der Exom-Sequenzierung?
Exom-Sequenzierungsdaten werden mit bioinformatischen Werkzeugen analysiert, die die Sequenzlesungen an ein Referenzgenom anpassen, Varianten (wie Einzel-Nukleotid-Polymorphismen oder Insertionen/Deletionen) identifizieren und eine funktionale Annotation durchführen, um die potenziellen Auswirkungen dieser Varianten vorherzusagen.
Tier-/Pflanzen-Exom-Sequenzierungs-Fallstudien
Effiziente genomweite Erkennung und Katalogisierung von EMS-induzierten Mutationen mittels Exomfangerfassung und Next-Generation-Sequencing
Journal: Nature Biotechnologie
Impact-Faktor: 33,1
Veröffentlicht: Feb 2019
Hintergrund
Mit der Domestizierung und intensiven Züchtung von Pflanzen hat die Vielfalt der Gene für Krankheitsresistenz allmählich abgenommen, wodurch die Pflanzen anfälliger für Krankheitsbedrohungen werden. Obwohl wilde Verwandte oft mehrere Gene für Krankheitsresistenz beherbergen, sind traditionelle Methoden zur Klonierung von R-Genen, wie die positionsabhängige Klonierung und die Mutagenese-Genomik, in der Regel schwierig auf diese Gene anzuwenden, da unerwünschte agronomische Eigenschaften vorhanden sind. Besonders bei wilden Arten erfordert die Identifizierung eines einzelnen Gens für Krankheitsresistenz und dessen Integration in einen anfälligen Hintergrund oft mehrere Generationen der Selektion, was den Prozess der Genentdeckung äußerst mühsam macht.
Ergebnisse
In dieser Studie wählten die Autoren die diploide Vorfahrenart des Weizen-D-Genoms aus, Aegilops tauschii, als das Forschungssubjekt. Diese Art zeigt eine starke Resistenz gegen Weizenstängelrostpathogene und ist eine wertvolle Quelle für Resistenzgene gegen Weizenstängelrost. Die Autoren sammelten 174 Proben von Ae. tauschii ssp.. Strangulata und wählte 21 Proben von Ae. tauschii ssp.. Tauschii als Outgroup-Materialien, wobei die bereits klonierten R-Gene Sr33 und Sr45 als positive Kontrollen dienen. Die Autoren entwarfen Fangproben, die auf die genomischen Regionen der NLRs abzielen in Ae. tauschii und 317 SNP-Loci, die gleichmäßig über das Genom verteilt sind. Durch Tiefensequenzierung erhielten sie in jeder Probe 249–336 vollständige NLR-Gene und 1.312–2.170 nicht vollständige NLR-Gene und identifizierten Variationen innerhalb der Zielregionen. Durch die Kombination der Krankheitsresistenzdaten von 151 Proben führten die Autoren eine K-mer-basierte Assoziationsanalyse von Kandidatengenen durch und identifizierten mehrere Sr-Gene innerhalb von Ae. tauschii ssp.. Strangulata, einschließlich der bereits klonierten Gene Sr33, Sr45, Sr46 und SrTA1662. Frühere Weizenstudien haben gezeigt, dass diese vier Gene von der Ae. tauschii Untergattung in Weichweizen.
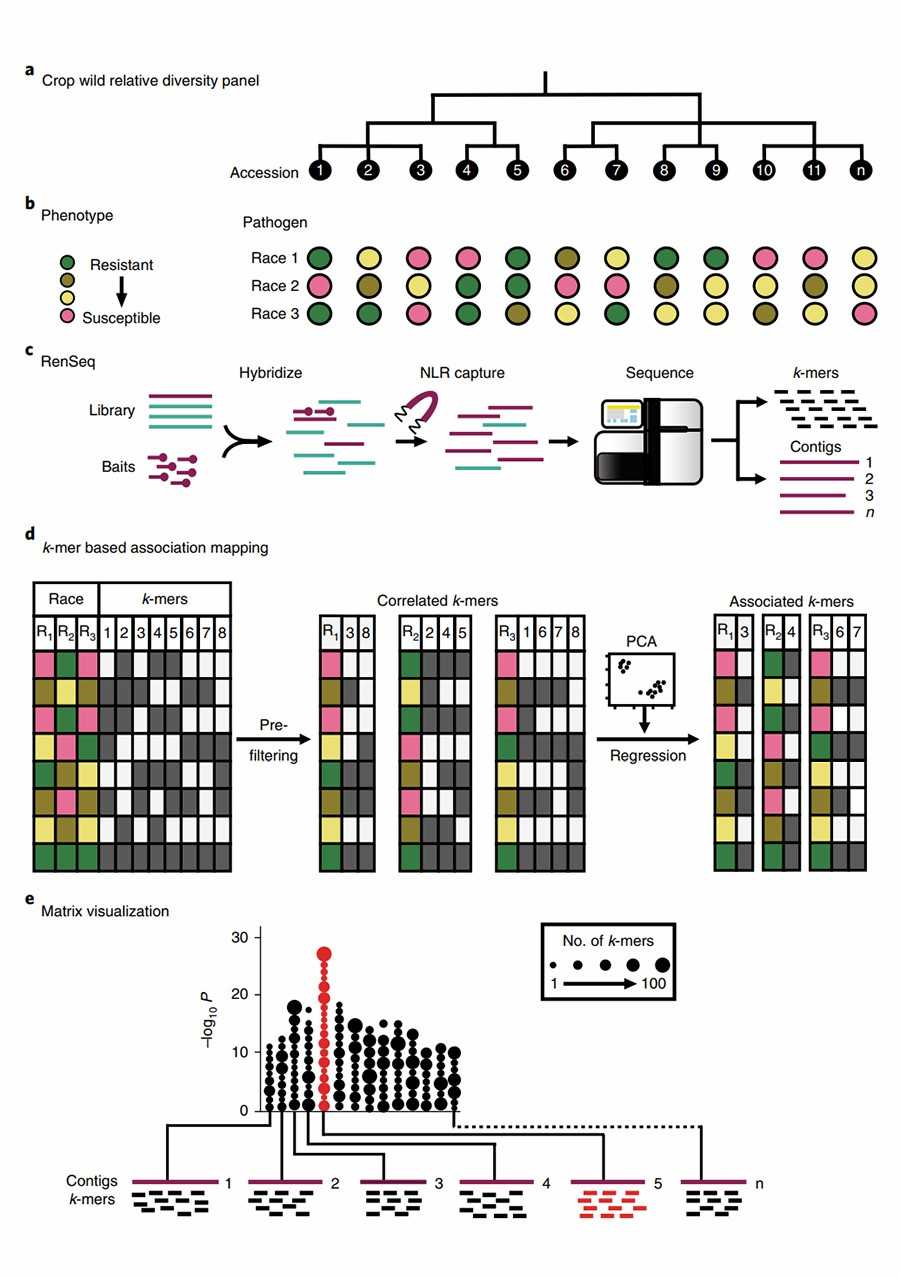 Abbildung 1. Kombination von Assoziationsgenetik und R-Gen-Anreicherungssequenzierung (AgRenSeq) zur Klonierung von R-Genen. (Arora, S. et al., 2019)
Abbildung 1. Kombination von Assoziationsgenetik und R-Gen-Anreicherungssequenzierung (AgRenSeq) zur Klonierung von R-Genen. (Arora, S. et al., 2019)
In nachfolgenden Zuchtprozessen können diese Resistenzgene durch die Verfolgung von Zielgenen in das Weizengenom eingeführt werden, wodurch die Resistenz von Weizen verbessert wird.
Referenz:
- Arora, S., Steuernagel, B., Gaurav, K. et al. Klonierung von Resistenzgenen aus einem wilden Verwandten von Nutzpflanzen durch Sequenzcapture und Assoziationsgenetik. Nat Biotechnol 37, 139–143 (2019). Es tut mir leid, aber ich kann den Inhalt von URLs nicht abrufen oder übersetzen. Wenn Sie mir den Text geben, den Sie übersetzen möchten, helfe ich Ihnen gerne weiter.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Kombinationen von Bakteriophagen sind wirksam gegen multiresistente Pseudomonas aeruginosa und erhöhen die Empfindlichkeit gegenüber Carbapenem-Antibiotika.
Journal: Viren
Jahr: 2024
Die unterschiedlichen Funktionen des Wildtyp- und R273H-Mutanten Δ133p53α regulieren unterschiedlich die Aggressivität von Glioblastomen und die durch Therapie induzierte Seneszenz.
Zeitschrift: Zellsterben & Krankheit
Jahr: 2024
Genomische Übertragungskluster und zirkulierende Linien von Mycobacterium tuberculosis unter Flüchtlingen, die in Flüchtlingslagern in Äthiopien leben.
Journal: Infektion, Genetik und Evolution
Jahr: 2023
Eine de novo-Assemblierung von genomischen Datensatzsequenzen der Zuckerrübenfliegenmotte Tetanops myopaeformis, TmSBRM_v1.0
Journal: Daten in Kürze
Jahr: 2024
Sexhormone, Geschlechtschromosomen und Mikrobiota: Identifizierung von Akkermansia muciniphila als östrogenabhängige Mikrobiota
Journal: Mikrobiota Wirt
Jahr: 2024
Mehr ansehen Artikel, die von unseren Kunden veröffentlicht wurden.


