
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Dual-RNA-Seq
Dual-RNA-Seq hat sich als Möglichkeit erwiesen, alle Klassen von kodierenden und nicht-kodierenden Transkripten sowohl des Wirts als auch des Erregers gleichzeitig zu überwachen. CD Genomics bietet einen hochauflösenden, erschwinglichen und unkomplizierten Dual-RNA-Seq-Service an, um direkte Einblicke in das Zusammenspiel von Wirt und Erreger zu ermöglichen.
Was ist Dual-RNA-Seq?
Es gibt eine Vielzahl von Wechselwirkungen zwischen Arten, wie Parasitismus, Symbiose, Konkurrenz usw. Die herkömmliche Transkriptom-Sequenzierung kann nur die Informationen einer einzelnen Art untersuchen, was nicht nur einen Teil der Daten verschwendet, sondern auch das Probenmaterial selbst während der Trennung von zwei Arten beeinträchtigt.
Die Sequenzierungstechnologie Dual RNA-seq wurde genau entwickelt, um solche Anfragen zu beantworten. Durch den Aufbau nur einer Transkriptom-Bibliothek ermöglicht Dual RNA-seq von total gemischter RNA nach doppelter rRNA-Depletion oder Poly(A)-Capture die Sequenzierung und Analyse von zwei (oder mehr) Arten zur gleichen Zeit, ohne die Arten trennen zu müssen, und offenbart damit die dynamischen Veränderungen der Genexpression zwischen ihnen. Gleichzeitig wird durch das Interaktionsmodell-Diagramm die regulatorische Beziehung von Genen und der Interaktionsmechanismus zwischen zwei Arten erfasst; um das regulatorische Netzwerk im Interaktionsprozess zu untersuchen, den Mechanismus der Pathogeninfektion und die Wirtsresistenz gegen Krankheiten zu analysieren; und um die evolutionäre Beziehung von Pathogenen zwischen verschiedenen Arten zu untersuchen und weiter die positive Selektion verwandter Gene basierend auf homologen Genen zu erforschen.
CD Genomics kann mit verschiedenen Invasionsmodellen umgehen - Pathogene, die Bakterien, Pilze, Protozoen usw. betreffen, der Wirt kann ein Säugetier oder eine Pflanze sein. Unser Ziel ist es, umfassende Dual-RNA-Seq-Dienstleistungen von der Versuchsplanung bis zur biokomputationalen Analyse anzubieten, um Ihre Forschungsbedürfnisse zu unterstützen.
Vorteile unseres Dual RNA-seq-Services
- Verfügbar für verschiedene Invasionsmodelle
- Flexibilität des Proben Typs: total gemischte RNA, infizierte Wirtszellen usw.
- Die Technologie der molekularen Barcodes ermöglicht kleine Mengen an Eingangs-Templates.
- Umfassende Analyse, hohe Stabilität: Nutzung der fortschrittlichen Illumina-Plattform-Sequenzierung in Kombination mit Hochleistungsrechnen (HPC), um eine schnelle und stabile Analyse und Bereitstellung von Sequenzierungsdaten zu erreichen.
- Strenge Qualitätskontrolle, vielfältige Analyse-Pipelines: Implementierung strenger Qualitätskontrollmaßnahmen und Angebot vielfältiger Analyse-Pipelines. Nicht nur Bereitstellung von Transkriptomanalysen für Arten, sondern auch Konfiguration fortgeschrittener Analysen wie Weighted Gene Co-expression Network Analysis (WGCNA), Annotation von Virulenzfaktoren, Analyse von Protein-Protein-Interaktionsnetzwerken usw., um Informationen über interspezies Beziehungen tiefgehend zu erkunden.
- Wissenschaftliches und sorgfältiges Design: Von der Probenauswahl über die Bibliotheksvorbereitung, Sequenzierung bis hin zur Datenanalyse durchläuft jeder Schritt ein wissenschaftliches und sorgfältiges Design, um qualitativ hochwertige Forschungsergebnisse zu gewährleisten.
Anwendungen von Dual-RNA-Seq
- Pflanzenkrankheiten und Mechanismen der Resistenz-Anfälligkeit.
- Physiologische Anpassung und molekulare Reaktionen.
- Pathogene und Wirtsimmunität.
- Interspezies-Symbiose und evolutionäre Mechanismen.
- Parasitismus in Schädlingen und Wirtsabwehr.
- Kokultivierung von Bakterien/Fungi.
- Forschung zu Infektionskrankheiten.
- Vergleichende Genomik und pathogenetische Mechanismen.
Workflow von Dual-RNA-Seq
Bei der Vorbereitung Ihrer Probe bietet CD Genomics eine umfassende Palette von Sequenzierungs- und bioinformatischen Analyse-Dienstleistungen an.

Dienstspezifikation
Musteranforderungen:
|
|
 |
Sequenzierung:
|
 |
Datenanalyse
|
Analyse-Pipeline
 Generische Pipeline zur Analyse von Dual-RNA-seq-Daten (Abbildung von V. Arluison et al., 2018)
Generische Pipeline zur Analyse von Dual-RNA-seq-Daten (Abbildung von V. Arluison et al., 2018)
Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details im Dual-RNA-Seq für Ihre Schreibweise (Anpassung)
Dual-RNA-Sequenzierung (Dual RNA-seq) ist eine hochmoderne Sequenziertechnik, die entwickelt wurde, um solche Herausforderungen zu bewältigen. CD Genomics, spezialisiert auf die Untersuchung von Wirt-Pathogen-Interaktionen, nutzt eine fortschrittliche Analyse von Dual-RNA-seq-Daten, um wissenschaftliche Fortschritte in diesem Bereich voranzutreiben. Unsere maßgeschneiderten Lösungen, unsere Expertise in der Bioinformatik und unser unerschütterliches Engagement für Qualität gewährleisten, dass Forscher genaue, zuverlässige und bedeutungsvolle Einblicke in die komplexe Welt der Wirt-Pathogen-Interaktionen gewinnen. Für weitere Informationen über unsere Dienstleistungen laden wir Sie ein, mit uns in Kontakt zu treten.
Referenzen:
- Alexander J. Westermann et al., Dual-RNA-Seq enthüllt die Funktionen nicht-kodierender RNAs in Wirt-Pathogen-Interaktionen. Natur2016, Bd. 000.
- Alexander J. Westermann et al., Auflösung von Wirt-Pathogen-Interaktionen durch duales RNA-Sequencing. PLoS Pathog2017, 13(2).
- Véronique Arluison und Claudio Valverde (Hrsg.), Bakterielle regulatorische RNA: Methoden und Protokolle. Methoden in der Molekularbiologie2018, Bd. 1737.
- Pisu et al., Dual-RNA-Sequenzierung von Mtb-infizierten Makrophagen in vivo zeigt ontologisch unterschiedliche Wirt-Pathogen-Interaktionen. Zellberichte2020, Bd. 30.
- Nuss A M, Beckstette M, Pimenova M, et al. Die Gewebe-Dual-RNA-Seq ermöglicht eine schnelle Entdeckung von infektion-spezifischen Funktionen und Riboregulatoren, die die Transkriptome von Wirt und Pathogen formen. Mitteilungen der Nationalen Akademie der Wissenschaften, 2017, 114(5): E791-E800.
Demo-Ergebnisse
Teilweise Ergebnisse sind unten aufgeführt:

Sequenzierungsqualitätsverteilung
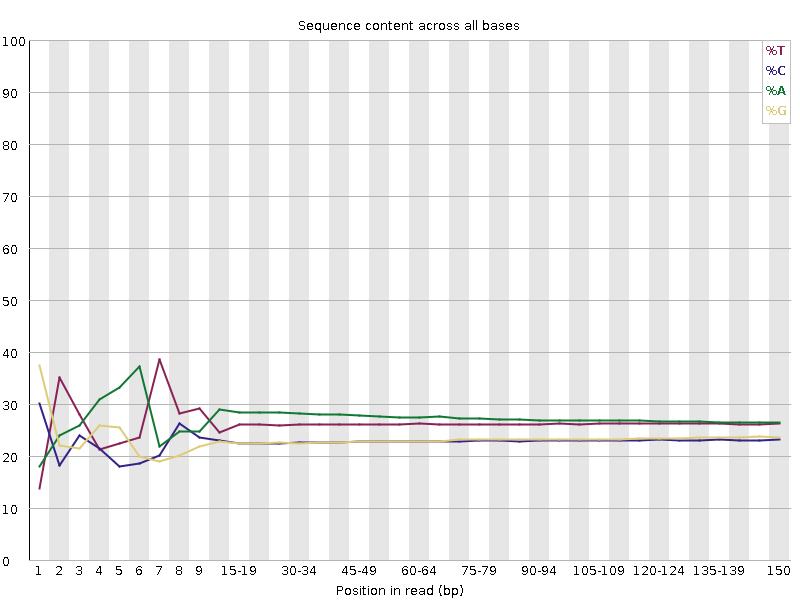
A/T/G/C-Verteilung
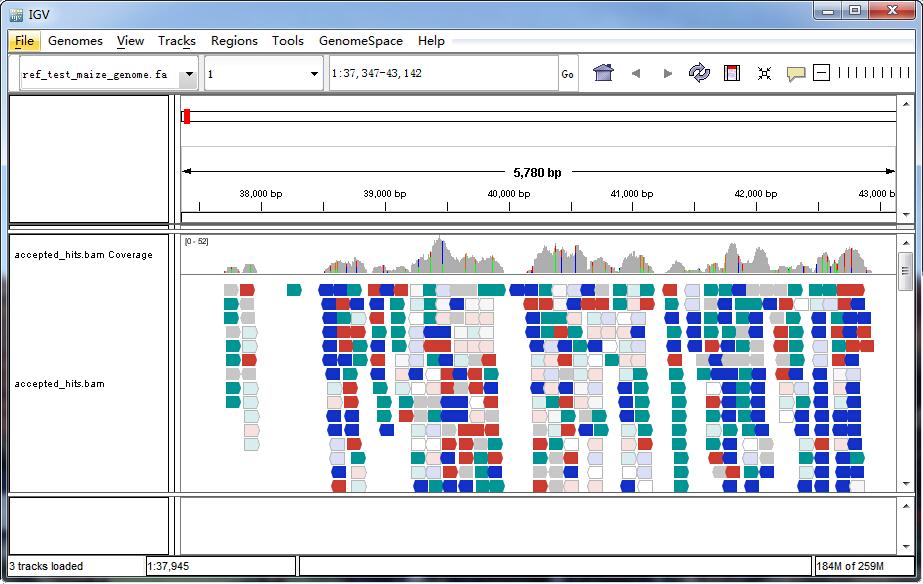
IGV-Browser-Oberfläche
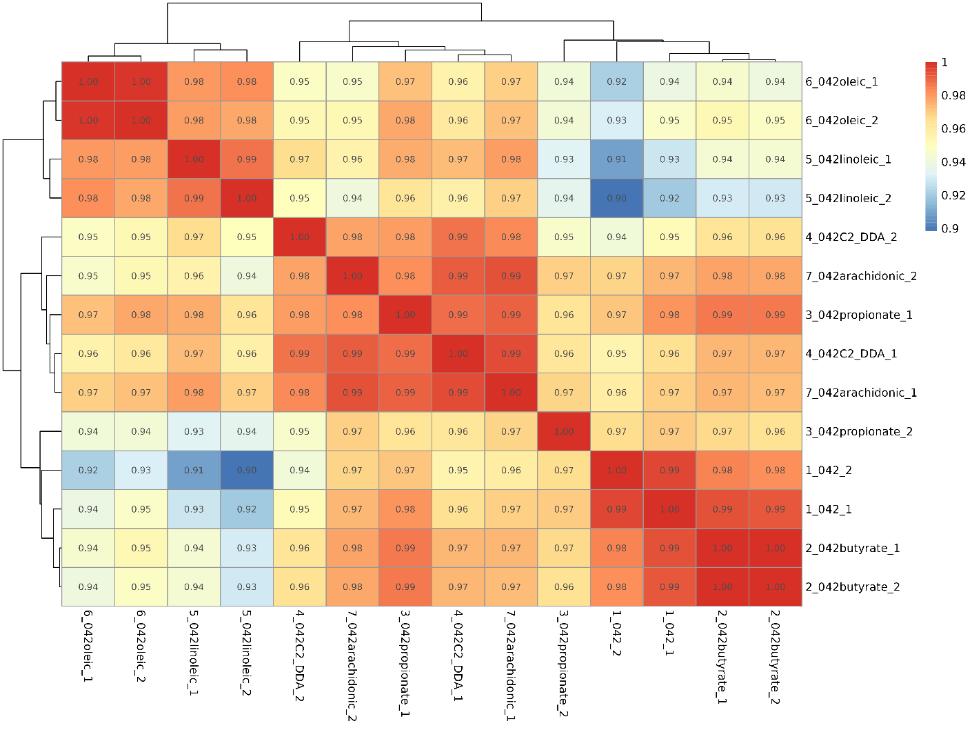
Korrelationsanalyse zwischen Proben
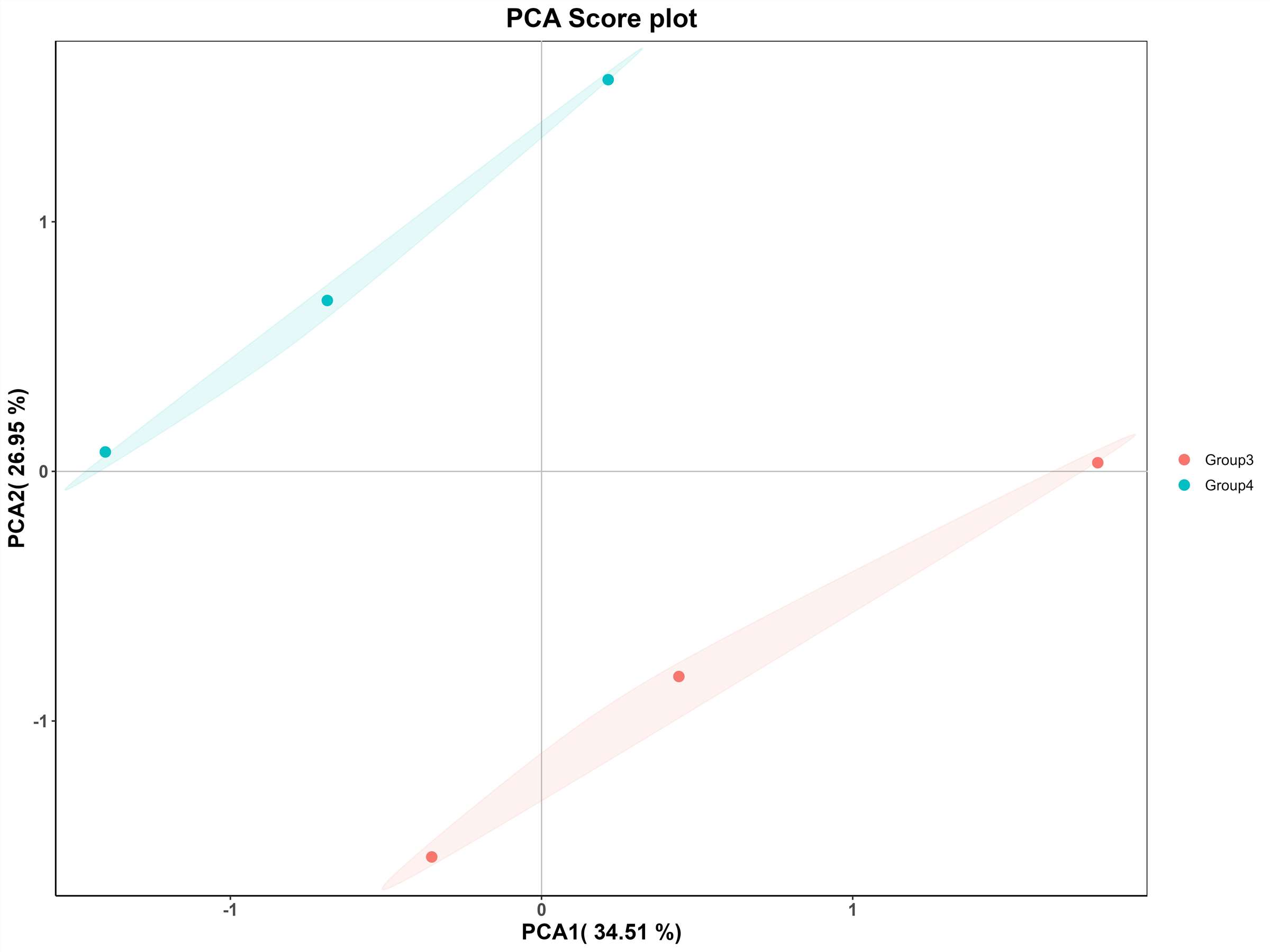
PCA-Score-Diagramm

Venn-Diagramm
Vulkan-Plot
Statistische Ergebnisse der GO-Annotation
KEGG-Klassifikation
Häufig gestellte Fragen zu Dual RNA-seq
1. Sind Referenzgenome für alle Arten erforderlich, die Dual-RNA-Seq durchlaufen?
Idealerweise würde die Verfügbarkeit von Referenzgenomen für beide interagierenden Arten genauere Ergebnisse ermöglichen. Die Literatur dokumentiert jedoch auch Verfahren, bei denen nur eine einzige Art ein Referenzgenom zur Verfügung hat. Der Analyseworkflow in solchen Fällen besteht zunächst darin, die Sequenzierungsdaten auf die Art mit einem Referenzgenom abzubilden. Die transkriptomischen Daten, nach dem Ausschluss der Mapping-Daten, können für die Informationen der anderen Art durch Mapping mit einem nahen Verwandten oder durch eine de novo-Assemblierung untersucht werden, was nachfolgende Analysen erleichtert. Insbesondere für den prokaryotischen Abschnitt in einer interagierenden Probe ist das Vorhandensein eines Referenzgenoms unerlässlich. In dessen Abwesenheit kann jedoch das bakterielle 'Pan-Genom'-Profiling implementiert werden.
2. Was sind die Vorteile von interaktiven Transkriptomen im Vergleich zu gewöhnlichen Transkriptomen?
Dual-RNA-Seq ist eine spezialisierte Sequenzierungstechnik, die entwickelt wurde, um Interaktionen zwischen verschiedenen Arten zu untersuchen, die im Allgemeinen symbiotische, parasitäre, wettbewerbsfähige und räuberische Beziehungen umfassen. Traditionelle Transkriptomik notwendig, die Trennung dieser interagierenden Arten vorzunehmen, ein Ansatz, der zwar nützlich ist, jedoch zu einem Verlust relevanter Informationen führen und potenzielle Artefakte im Zusammenhang mit dem Trennungsprozess einführen kann. Im krassen Gegensatz dazu steht der duale RNA-seq-Ansatz, der gleichzeitige Untersuchungen von zwei oder mehreren interagierenden Arten ermöglicht, ohne dass eine Trennung erforderlich ist. Dadurch wird der mögliche nachteilige Einfluss, der durch die Trennung der Arten entstehen könnte, umgangen. Darüber hinaus ist diese Methodik kosteneffektiv, da nur eine Transkriptom-Bibliothek erstellt werden muss, die eine gleichzeitige Analyse aller einbezogenen Arten ermöglicht.
3. Was sind die verschiedenen Arten von RNA-Seq?
Darüber hinaus kann RNA-Seq je nach Forschungszielen und Besonderheiten der Probe in mehrere Typen unterteilt werden, wie zum Beispiel: Gesamt-RNA-Sequenzierung, mRNA-Seq, miRNA-seq, Lange nicht-kodierende RNA-seq, Vollständige Transkriptom-Sequenzierung, Zirkuläre RNA-Sequenzierungund Ganz Exom RNA-Seq.
Dual-RNA-Seq-Fallstudien
Dual-RNA-Seq deckt die Abhängigkeit von metabolischen Aminosäuren des intrazellulären Bakteriums Piscirickettsia salmonis auf, das atlantischen Lachs infiziert.
Zeitschrift: Frontiers in Mikrobiologie
Impactfaktor: 6,064
Veröffentlicht: 27. November 2018
Zusammenfassung
Hochdurchsatz-Sequenzierung Technologien, die in der transkriptomischen Forschung angewendet werdenRNA-Seq), haben die Möglichkeit eröffnet, komplexe molekulare Reaktionen in verschiedenen biologischen Kontexten zu verstehen. Kürzlich hat sich das synchrone Sequenzieren der Transkriptome eines Pathogens und des Wirts während der Infektion – bekannt als Dual-RNA-Seq – als vielversprechende Methode zur Enthüllung der Feinheiten der Wirt-Pathogen-Interaktionen herausgestellt. In dieser speziellen Studie analysierten die Forscher durch duale rRNA-Depletion und RNA-Sequenzierungsansatz gleichzeitig die Transkriptome des intrazellulären Bakteriums. Piscirickettsia bei Lachsen und ihren W Wirtsarten, Atlantischer Lachswährend des Verlaufs der Infektion.
Methoden
- Kopfniere und Milzgewebe
- Totale RNA-Isolierung
- Entfernen Sie sowohl bakterielle als auch Wirts-rRNAs.
- Dual-RNA-Seq
- Illumina HiSeq-Plattform
- 100 bp gepaarte Endlesungen
- Differenzielle Expressionsanalyse
- Funktionale Annotation
- Gene-Ontologie (GO) Annotation
- KEGG-Pfadannotationsanalyse
- qPCR-Validierungen
Ergebnisse
Erforschung des Transkriptoms von Wirt und Pathogen während der Pathogenese
Die Dual-RNA-Seq-Analyse delineierte die Regulation der Transkriptome von P. salmonis und S. salar im Verlauf der Infektion. Bezüglich des Transkriptoms von S. salar wurden 771 und 829 Gene in der Milz bei 7 bzw. 14 dpi unterschiedlich exprimiert (Abb. 1B). Gleichzeitig wurden 412 und 467 Gene zu denselben Zeitpunkten in der Kopfniere unterschiedlich reguliert. Speziell in der Kopfniere modulierte Gene umfassten solche, die für das MATE-Effluxfamilienprotein, das DNA-Reparaturprotein RecN, das Chaperonprotein HtpG und verschiedene Membranproteine kodieren (Abb. 2). Unter den gemeinsamen Genen wurden verschiedene Proteinphosphatasen 1, ribosomale Proteine, molekulare Chaperone und Asn/Gln-Transamidosom-Untereinheiten identifiziert (Abb. 2).
 ABBILDUNG 1. (A) Globale Transkriptomanalyse von Piscirickettsia salmonis (blau) und S. salar (rot) bei 3, 7 und 14 Tagen nach der Infektion (dpi) in Milz- und Kopfniere-Geweben. Die Expressionswerte wurden als Transkripte pro Million Reads (TPM) geschätzt und für die Heatmap-Clustering Log2-transformiert. (B) Differenziell exprimierte Gene in Milz und Kopfniere bei 7 und 14 dpi unter Verwendung der Expressionswerte bei 3 dpi als Kontrolle. Drei Tage nach der Infektion wurden als Basislinie für die Genexpression verwendet, da in keiner Art von Kontrollgruppe bakterielle Reads vorhanden sein würden.
ABBILDUNG 1. (A) Globale Transkriptomanalyse von Piscirickettsia salmonis (blau) und S. salar (rot) bei 3, 7 und 14 Tagen nach der Infektion (dpi) in Milz- und Kopfniere-Geweben. Die Expressionswerte wurden als Transkripte pro Million Reads (TPM) geschätzt und für die Heatmap-Clustering Log2-transformiert. (B) Differenziell exprimierte Gene in Milz und Kopfniere bei 7 und 14 dpi unter Verwendung der Expressionswerte bei 3 dpi als Kontrolle. Drei Tage nach der Infektion wurden als Basislinie für die Genexpression verwendet, da in keiner Art von Kontrollgruppe bakterielle Reads vorhanden sein würden.
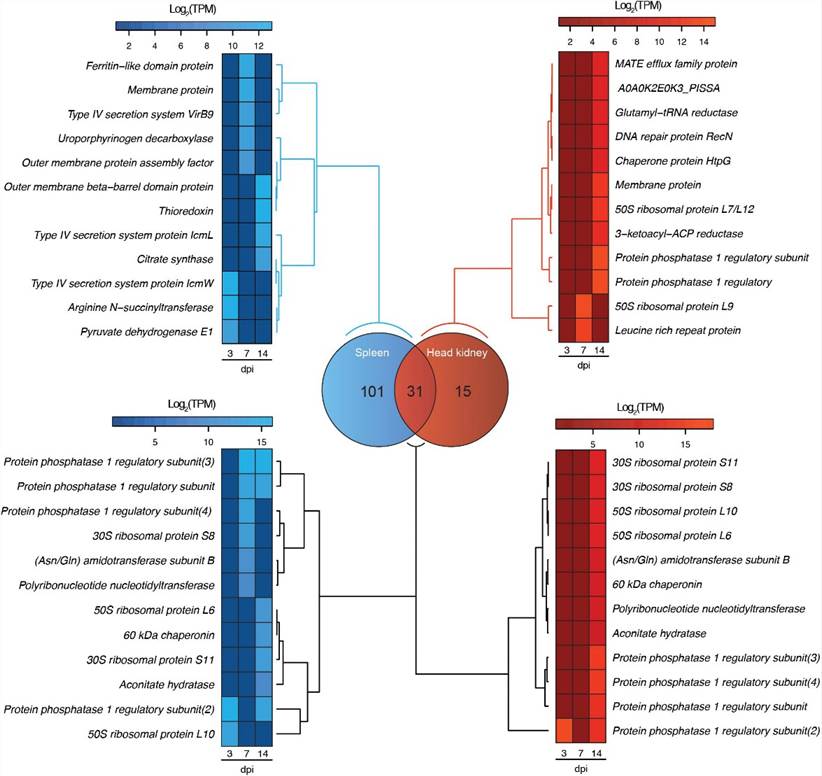 ABBILDUNG 2. Venn-Diagramm, das die Anzahl der exklusiven und gemeinsamen Gene im Transkriptom von zeigt. P. salmonis während der Infektion in der Milz (blau) und den Kopfniere (rot) Geweben von Atlantischem Lachs. Die Heatmaps zeigen eine Untergruppe von exklusiven und gemeinsamen Genen, die exprimiert werden von P. salmonis in beiden Geweben.
ABBILDUNG 2. Venn-Diagramm, das die Anzahl der exklusiven und gemeinsamen Gene im Transkriptom von zeigt. P. salmonis während der Infektion in der Milz (blau) und den Kopfniere (rot) Geweben von Atlantischem Lachs. Die Heatmaps zeigen eine Untergruppe von exklusiven und gemeinsamen Genen, die exprimiert werden von P. salmonis in beiden Geweben.
Aminosäurestoffwechsel: Eine gemeinsame Reaktion zwischen P. salmonis und S. salar
Die GO-Anreicherungsanalyse zeigte, dass ein großer Anteil der differentially exprimierten Gene in P. salmonis waren mit den Stoffwechselprozessen von Proteinen und Stickstoffverbindungen assoziiert. Eine Anreicherung von Genen, die mit dem Proteinmetabolismus in Verbindung stehen, wurde ebenfalls durch KEGG-Annotation entdeckt. Allerdings, obwohl der Aminosäurestoffwechsel nicht die Haupttranskriptomantwort im Wirt darstellt, sowohl P. salmonis und S. salar zahlreiche Gene nachgewiesen, die mit der biologischen Synthese und dem Abbau von Aminosäuren verbunden sind.
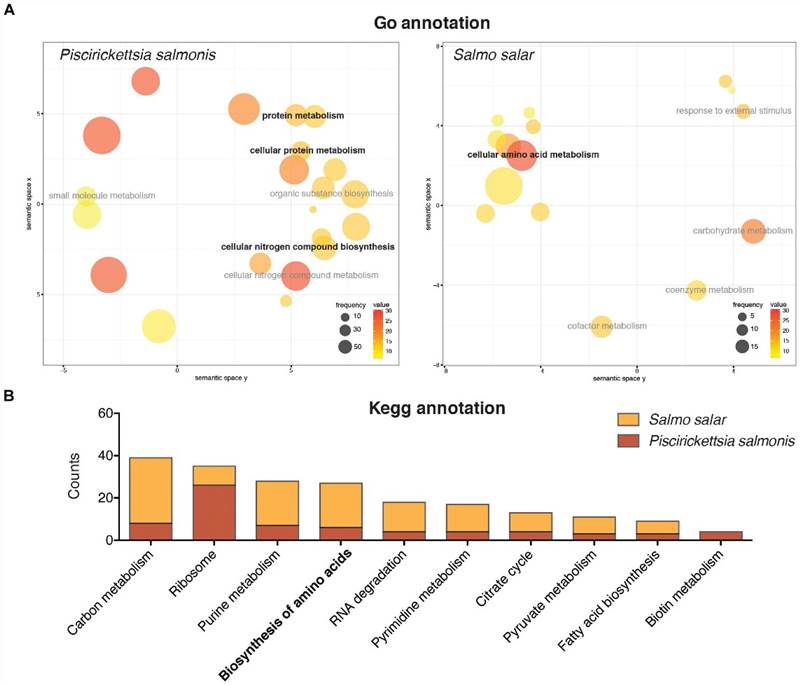 ABBILDUNG 3. Genontologie-Anmerkung (A) und KEGG-Pfad-Anmerkung (B) der differentially exprimierten Gene in Atlantischer Lachs und P. salmonis während der Infektion.
ABBILDUNG 3. Genontologie-Anmerkung (A) und KEGG-Pfad-Anmerkung (B) der differentially exprimierten Gene in Atlantischer Lachs und P. salmonis während der Infektion.
Fazit
Über die standardmäßigen Immunantworten des Wirts und die Virulenzfaktoren von Krankheitserregern hinaus zeigen sowohl Bakterien als auch der Wirt eine Fülle von Genen, die mit Stoffwechselprozessen assoziiert sind, insbesondere mit dem Aminosäurestoffwechsel. Unsere Ergebnisse deuten auf eine Abhängigkeit von P. salmonis über den Aminosäurestoffwechsel von S. salar, was auf einen neuartigen Pathogenese-Mechanismus hindeuten könnte, der auf der Fähigkeit basiert, Nährstoffe vom Wirt aufzunehmen. Im weitesten Sinne ermöglicht die duale Transkriptom-Sequenzierung eine vielfältige Perspektive auf die Interaktion zwischen Wirt und Pathogen, die über die biologischen Prozesse hinausgeht, die mit der Immunität verbunden sind.
Referenz:
- Valenzuela-Miranda D, Gallardo-Escárate C. Dual-RNA-Seq enthüllt die metabolische Abhängigkeit von Aminosäuren des intrazellulären Bakteriums Piscirickettsia salmonis, das Atlantischen Lachs infiziert. Grenzen der Mikrobiologie, 2018, 9: 418416.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Chaperon-vermittelte Autophagie steuert proteomische und transkriptomische Wege, um die Aktivität von Gliom-Stammzellen aufrechtzuerhalten.
Journal: Krebsforschung
Jahr: 2022
Zirkuläre DNA-Tumorviren erzeugen zirkuläre RNAs.
Zeitschrift: Mitteilungen der Nationalen Akademie der Wissenschaften
Jahr: 2018
Wiederholte Immunisierung mit ATRA-haltigem liposomalem Adjuvans transdifferenziert Th17-Zellen zu einem Tr1-ähnlichen Phänotyp.
Zeitschrift: Zeitschrift für Autoimmunität
Jahr: 2024
Die Rolle der Histonvariante H2A.Z.1 in Gedächtnis, Transkription und alternativer Spleißung wird durch Lysinmodifikation vermittelt.
Zeitschrift: Neuropsychopharmakologie
Jahr: 2024
FAK-Verlust reduziert die ERK-Phosphorylierung, die durch BRAFV600E induziert wird, um die intestinale Stammzell-Eigenschaft und die Bildung von Blinddarmtumoren zu fördern.
Journal: Elife
Jahr: 2023
Identifizierung von zirkulären RNAs, die die Proliferation von Kardiomyozyten in neugeborenen Schweineherzen regulieren
Journal: JCI Insight
Jahr: 2024
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


