
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
microRNA-Sequenzierungsdienst
Was ist die MikrorNA-Sequenzierung?
MicroRNA (miRNA) ist eine Klasse von endogenen kleinen nicht-kodierenden RNAs in Eukaryoten, mit einer Länge von etwa 17 bis 25 Basenpaaren. Seit der Entdeckung von miRNA im Jahr 1993 haben Fortschritte und Entdeckungen im Bereich der kleinen RNAs das Verständnis der Genregulation in der wissenschaftlichen Gemeinschaft revolutioniert. Von der embryonalen Entwicklung über die Zellapoptose bis hin zum Tumorwachstum spielt miRNA eine entscheidende Rolle in einer Vielzahl von physiologischen und pathologischen Prozessen. Verschiedene mit Genetik, Stoffwechsel, Infektionskrankheiten und Tumoren assoziierte miRNAs bieten Wissenschaftlern neue Perspektiven für die pathologische Forschung und könnten als zuverlässige Biomarker für Krankheiten dienen. Wissenschaftler erforschen aktiv Interventionen und therapeutische Behandlungen für Krankheiten, indem sie die Funktionen von miRNA manipulieren und neuartige in vivo-Übertragungsmethoden entwickeln. Darüber hinaus wurde entdeckt, dass miRNA die evolutionäre Anpassung und den Artenwechsel vermittelt, und es gibt noch viel mehr zu entdecken in Bezug auf miRNA.
miRNA-seq ist ein Hochdurchsatz-Sequenzierung Eine Methode wurde entwickelt, um die Expression von miRNAs nachzuweisen. Drosha und Dicer, die an der Verarbeitung von miRNAs beteiligt sind, gehören zur RNase III-Familie der Nukleasen. Daher haben miRNAs eine 5'-PO4-Gruppe und eine 3'-OH-Gruppe, die direkt für Ligationreaktionen verwendet werden können. Darüber hinaus sind miRNAs durch Längen von 18-30 Nukleotiden gekennzeichnet. Durch die direkte Ligation von Gesamt-RNA und die Rückgewinnung der eingefügten Fragmente mit Längen von 18 bis 30 Nukleotiden können miRNAs angereichert, Bibliotheken erstellt und die Expression von miRNAs analysiert werden.
Unser miRNA-Sequenzierungsdienst:
CD Genomics bewirbt sich Hochdurchsatz-Sequenzierung Technologie zur Sequenzierung von microRNA-Bibliotheken. Diese Technologie ermöglicht den Erwerb von Millionen von microRNA-Sequenzen in einem einzigen Durchlauf, was eine schnelle Identifizierung von bekannten und unbekannten microRNAs sowie deren Ausdrucksunterschieden in verschiedenen Arten, Geweben, Entwicklungsstadien und Krankheitszuständen ermöglicht. Darüber hinaus ist die Next-Generation-Sequenzierung Technologie hat bedeutende Anwendungen in der Erkennung von komplementären Strängen von Mikro-RNAs, der Bearbeitung von Mikro-RNAs, der Erkennung von Mikro-RNA-Isoformen, der Vorhersage neuer Mikro-RNAs und der Analyse von Zielgenen von Mikro-RNAs. Sie bietet ein leistungsstarkes Werkzeug zum Studium der Rolle von Mikro-RNAs in zellulären Prozessen und ihren biologischen Auswirkungen.
Die Mikrorna-Sequenzierungsdienste von CD Genomics umfassen die Quantifizierung von vom Kunden bereitgestellten Gesamt-RNA- oder Mikrorna-Proben, gefolgt von der Konstruktion der Mikrorna-Bibliothek, der Cluster-Generierung und der Sequenzierung. Der Mikrorna-Sequenzierungsdienst von CD Genomics bietet umfassende und integrierte Dienstleistungen von der Probenentnahme bis zur Datenanalyse. Der Dienst umfasst die Qualitätskontrolle der Probenentnahme, die Bibliotheksvorbereitung, die Sequenzierung, die bioinformatische Analyse und die Datenberichterstattung.
Vorteile unseres miRNA-Sequenzierungsdienstes
- Hohe Empfindlichkeit: Theoretisch in der Lage, eine einzelne Kopie von Mikro-RNA nachzuweisen.
- Hohe Präzision: In der Lage, Einzelbasenunterschiede in Mikro-RNA zu erkennen.
- Unbeeinflusst von vorherigen Informationsstörungen, in der Lage, bekannte Mikro-RNAs zu identifizieren und neue zu entdecken.
- Bewahrt richtungsabhängige Informationen für strangspezifische Expressionsanalysen.
- Verwendet Barcodes, um mehrere Proben in einem einzigen Durchlauf wirtschaftlich zu analysieren.
- Höchste Empfindlichkeit und niedrigste Verzerrung.
- Nutzen von molekularen Barcodes und digitalen Tags zur Korrektur von falsch-positiven Duplikaten, die durch PCR und Sequenzierung eingeführt wurden.
- Starkes Bioinformatik-Analyse-Team, das umfassende Analyseergebnisse liefert.
Anwendungen der miRNA-Sequenzierung
- Eigenständige Nutzung: Untersuchung des Ausdrucks von miRNAs während biologischer Prozesse.
- Eigenständige Nutzung: Erforschung von miRNA-Biomarkern in biologischen Flüssigkeiten wie Serum, Plasma und Exosomen.
- Kombinierte Anwendung mit mRNA-SeqKonstruktion von miRNA-mRNA-Regulationsnetzwerken während biologischer Prozesse.
- Kombinierte Anwendung mit circRNA-seqAufbau von miRNA-circRNA-Regulierungsnetzwerken.
- Multi-Omics-Integration: Aufbau von circRNA-miRNA-mRNA-Regulationsnetzwerken.
miRNA-Sequenzierungs-Workflow

Dienstspezifikation
Beispielanforderungen
|
|
 Klicken |
Sequenzierungsstrategien
|
 |
Datenanalyse Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an:
|
Analyse-Pipeline

Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details zur MikrorNA-Sequenzierung für Ihr Schreiben (Anpassung)
CD Genomics bietet spezialisierte MikrorNA-Sequenzierungsdienste an, die Qualitätskontrolle der Proben, Bibliothekskonstruktion, Tiefensequenzierung und umfassende Datenanalyse umfassen. Unser maßgeschneiderter Ansatz gewährleistet präzise Ergebnisse, die auf Ihre Forschungsbedürfnisse abgestimmt sind. Kontaktieren Sie uns für weitere Informationen, wie wir Ihre Forschungsziele unterstützen können.
Demo-Ergebnisse
Teilweise Ergebnisse sind unten aufgeführt:

Sequenzierungsqualitätsverteilung
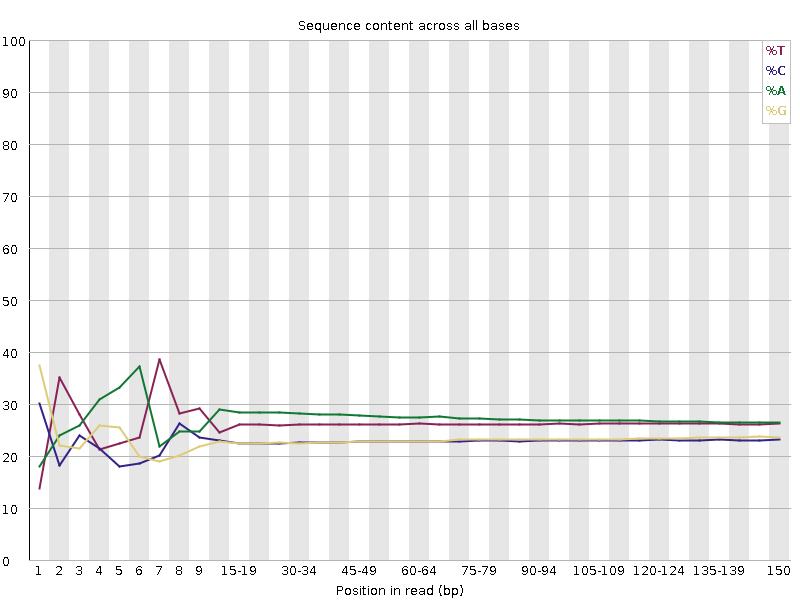
A/T/G/C-Verteilung
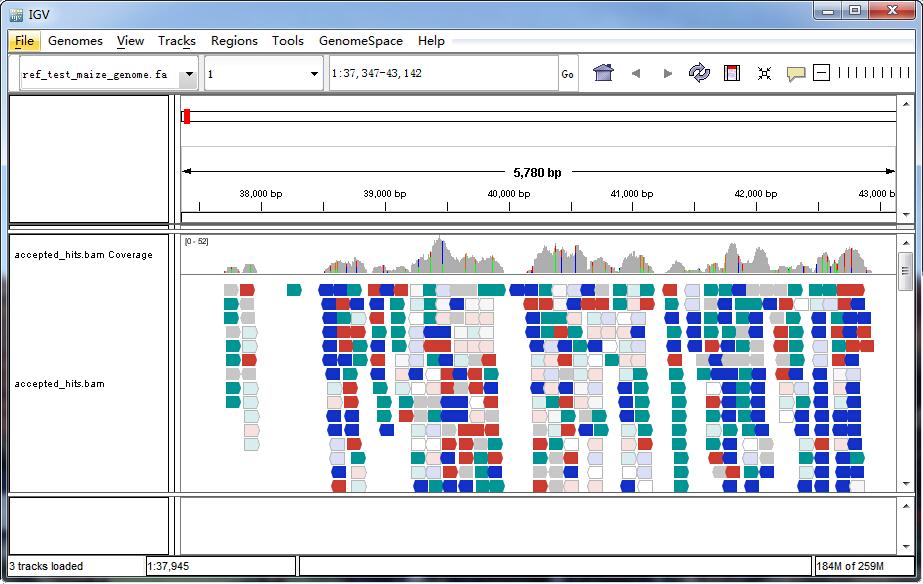
IGV-Browser-Oberfläche
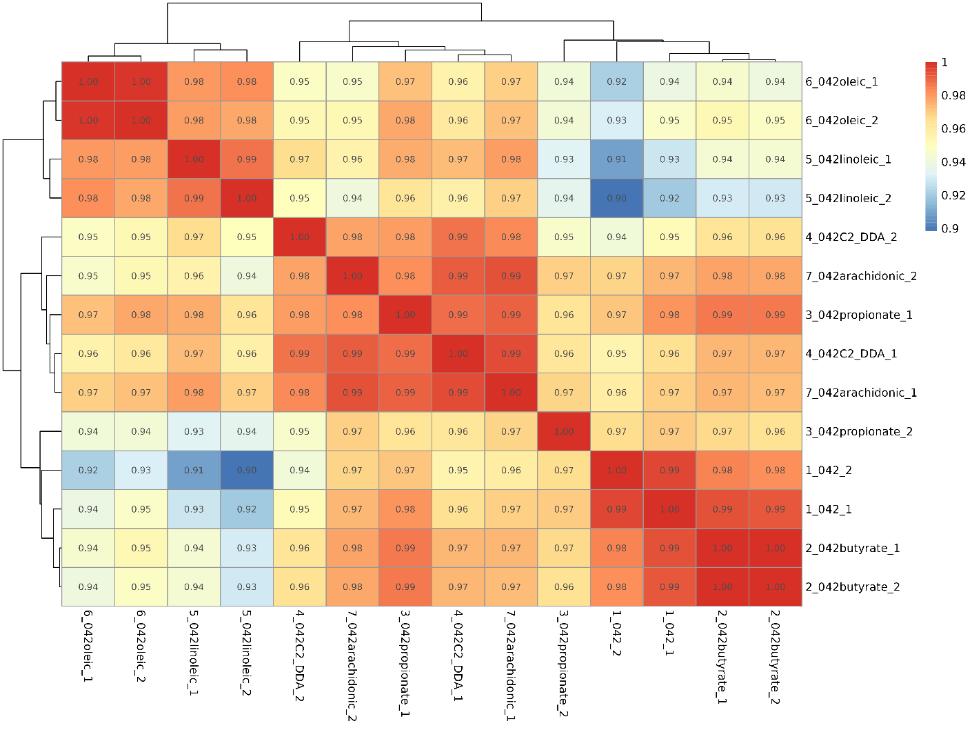
Korrelationsanalyse zwischen Proben
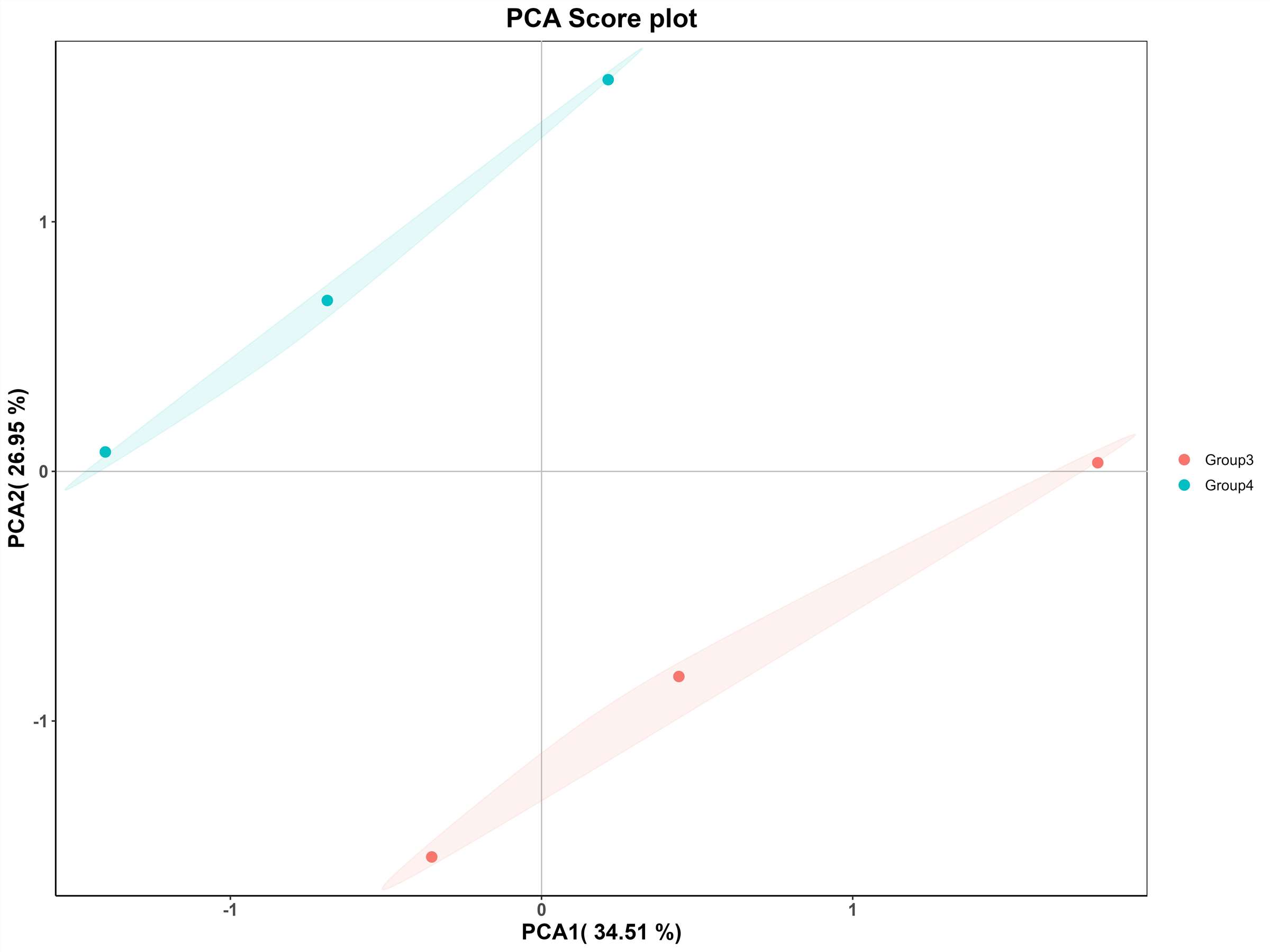
PCA-Score-Diagramm

Venn-Diagramm
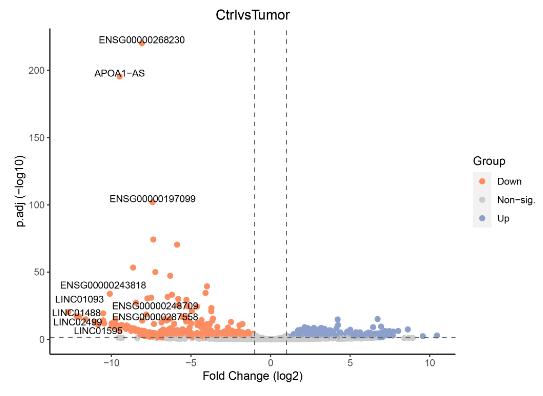
Volcano-Diagramm
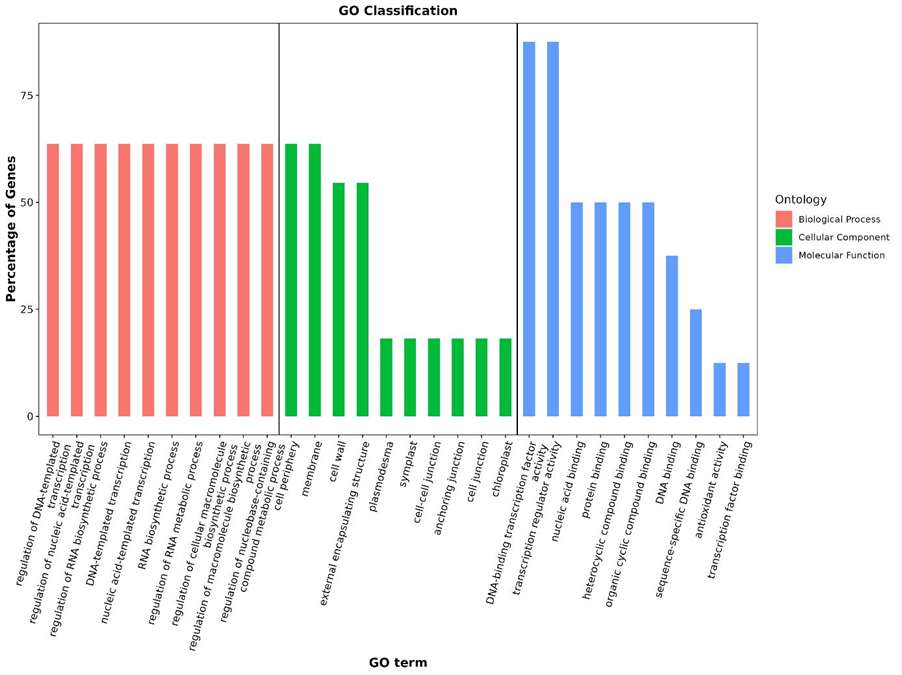
Statistik Ergebnisse der GO-Annotierung
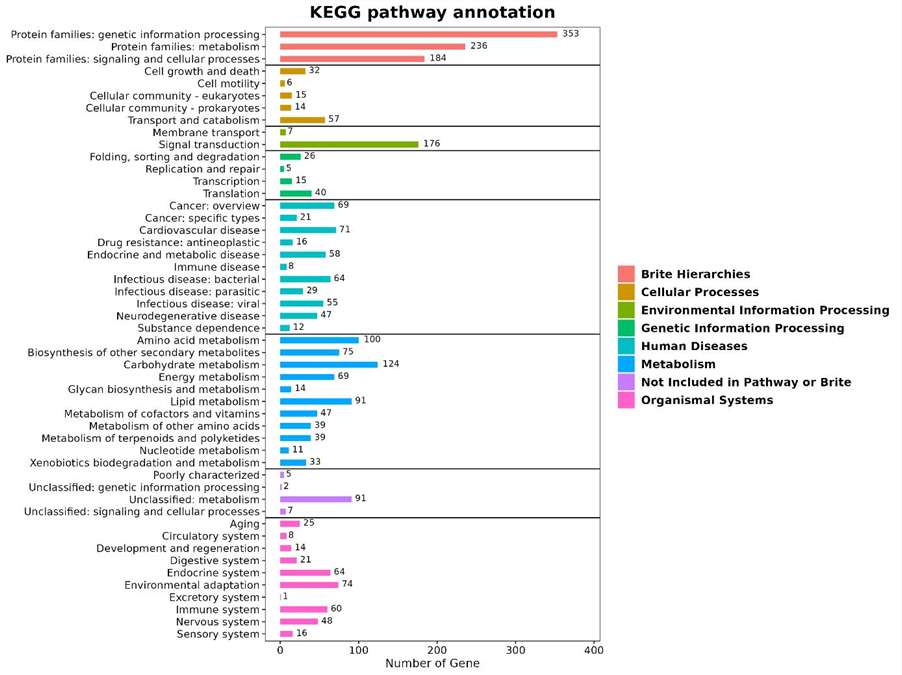
KEGG-Klassifikation
microRNA-Seq Häufig gestellte Fragen
1. Warum sich für die Durchführung von microRNA-Sequenzierung entscheiden?
Die MikrorNA-Sequenzierung ermöglicht eine umfassende Profilierung der Expressionsniveaus aller MikrorNAs in Zellen oder Geweben, einschließlich von niedermolekularen und seltenen MikrorNAs. Im Vergleich zu traditionellen Methoden wie RT-qPCR bietet sie eine höhere Sensitivität und Abdeckung. Dies ist entscheidend für das Studium der Zusammenhänge zwischen MikrorNAs und Krankheiten, biologischen Prozessen sowie Reaktionen auf Medikamente.
2. Was ist der Bibliothekskonstruktionsprozess der MikroRNA-Sequenzierung?
MicroRNA (miRNA) unterscheidet sich von messenger RNA (mRNA), langen nicht-kodierenden RNA (lncRNA) und zirkulärer RNA (circRNA) durch ihre kurzen Sequenzen. Daher werden während der Bibliothekskonstruktion die einzigartigen Eigenschaften der miRNA-Sequenzen direkt genutzt, indem Adapter hinzugefügt werden, gefolgt von reverser Transkription und Amplifikation. Anschließend werden Sequenzierungsadapter durch PCR-Amplifikation hinzugefügt, gefolgt von der Gelreinigung spezifischer DNA-Fragmente. Danach wird die Bibliothek maschinell sequenziert. Aufgrund des unterschiedlichen Prozesses der Bibliothekskonstruktion erfordert die gleichzeitige Detektion von miRNA und mRNA die Konstruktion separater Bibliotheken. Dies liegt daran, dass die Segmentauswahl im Bibliothekskonstruktionsprozess, die sich auf mRNA, lncRNA und circRNA bezieht, kleine RNA-Fragmente herausfiltert.
Was ist die miRNA-Saatsequenz?
Die Seed-Sequenz eines miRNA-Moleküls bezieht sich auf ein entscheidendes Segment, das typischerweise aus 2 bis 8 Nukleotiden besteht. Diese Sequenz befindet sich am 5'-Ende des MikroRNA-Moleküls und spielt eine zentrale Rolle bei der Bindung an die 3' UTR-Region der Zielgene. Durch komplementäres Pairing mit dem Zielgen über seine Seed-Sequenz moduliert die miRNA die Genexpression.
4. Wie lange dauert die MikroRNA-Sequenzierung?
Die Länge der microRNA-Sequenzierung variiert je nach verwendetem Sequenzierungsplattform und experimentellem Design. Im Allgemeinen liegen die erzeugten Reads in der microRNA-Sequenzierung typischerweise zwischen 18 und 50 Nukleotiden, abhängig von den technischen Einschränkungen des Sequenzierungsgeräts und den spezifischen Zielen des Sequenzierungsexperiments.
5. Welche Methoden gibt es zur Detektion von miRNA?
Die Nachweismethoden für miRNA umfassen Deep Sequencing, Mikroarray und Echtzeit-qPCR. Deep Sequencing ist eine hochmoderne Hochdurchsatz-Sequenzierung Technologie, die eine umfassende, tiefgehende und detaillierte Analyse eines biologischen Entität, Gewebes oder zellulären Bestandteils auf Genom-, Transkriptom- oder Metabolom-Ebene ermöglicht. Durch die Deep-Sequencing-Technologie können unbekannte miRNAs erforscht und entdeckt sowie bekannte miRNAs qualitativ und semi-quantitativ vorläufig analysiert werden. Mikroarray Die Technologie bietet Vorteile in Bezug auf hohe Durchsatzraten und Kosteneffizienz, hat jedoch begrenzte Möglichkeiten zur Differenzierung der Expression. Die Echtzeit-qPCR ist die empfindlichste und genaueste Methode zur Quantifizierung von miRNA zur Bestimmung der DNA- (oder cDNA-) Kopienzahlen in einer Probe.
6. Welche Vorteile bietet die MikrorNA-Sequenzierung im Vergleich zur traditionellen RT-qPCR?
Die MikrorNA-Sequenzierung bietet erhebliche Vorteile gegenüber der traditionellen RT-qPCR: Sie erkennt umfassend alle bekannten und potenziellen MikrorNAs, einschließlich solcher mit niedrigen Expressionsniveaus; sie ermöglicht eine Hochdurchsatzverarbeitung mehrerer Proben, wodurch die Arbeitseffizienz und Datenkonsistenz erhöht werden; zudem bietet sie detaillierte bioinformatische Analysen, einschließlich der Analyse der differentiellen Expression und der funktionalen Anreicherung, die ein tieferes Verständnis der Rollen und Mechanismen von MikrorNAs in biologischen Prozessen unterstützen.
microRNA-Seq Fallstudien
miRNA-Biomarker zur Vorhersage der Gesamtüberlebensraten bei Plattenepithelkarzinomen im Kopf-Hals-Bereich
Journal: Genomik
Impactfaktor: 6,205
Veröffentlicht: 2020
Hintergrund
Kopf- und Hals-Plattenepithelkarzinom (HNSCC) ist ein bösartiger Tumor des oberen Verdauungstrakts. Veränderungen in der Funktion von miRNA können die Krebsentwicklung durch verschiedene Mechanismen fördern. Diese Studie nutzte RNA-Sequenzierung (RNA-Seq) und miRNA-Sequenzierungsdaten aus der TCGA-Datenbank, um die dysregulierte miRNA-Mikroumgebung zu demonstrieren und wertvolle Biomarker für die miRNA-Therapie bereitzustellen. Im Trainingssatz wurden sieben miRNAs als unabhängige prognostische Faktoren für HNSCC-Patienten identifiziert. Diese miRNAs zielten gemeinsam auf 60 Gene ab, von denen neun Zielgene (CDCA4, CXCL14, FLNC, KLF7, NBEAL2, P4HA1, PFKM, PFN2 und SEPPINE1) mit dem Gesamtüberleben (OS) der Patienten assoziiert waren. Diese Studie identifizierte neuartige miRNA-Marker zur Prognosevorhersage von Plattenepithelkarzinomen im Kopf-Hals-Bereich.
Materialien & Methoden
- 529 (42 normale und 487 Tumor) Patienten
- 552 (44 normale und 508 Tumor) Patienten
- RNA-Seq
- miRNAs-seq
- Anreicherungsanalyse der Zielgene von miRNAs
- Standardisierte mRNA- und miRNA-Expressionsprofile
Ergebnisse
1. Datenerfassung
RNA-seq-Daten und miRNA-seq-Daten von HNSCC-Patienten wurden aus der TCGA-Datenbank heruntergeladen. Die miRBase-Datenbank enthält bekannte miRNA-Sequenzen und Annotationen, einschließlich Informationen über den Standort und die Sequenz reifer miRNAs. Die Fasta-Format-Sequenzen aller reifen miRNA-Sequenzen, die aus miRBase erhalten wurden, wurden für die miRNA-Annotation verwendet. RNA-seq-Daten und miRNA-seq-Daten wurden aus Proben von 529 Individuen (42 normal und 487 Tumor) bzw. 552 Individuen (44 normal und 508 Tumor) extrahiert. Die gleichzeitig gesammelten klinischen und pathologischen Daten umfassten Geschlecht, Alter, Staging, TNM-Klassifikation, Überlebensstatus und Überlebensdauer.
2. Etablierung von miRNA-basierten Prognosen
Marker Insgesamt wurden 1.075 unterschiedlich exprimierte mRNAs (581 hochreguliert und 494 herunterreguliert) sowie 313 unterschiedlich exprimierte miRNAs (203 hochreguliert und 110 herunterreguliert) aus der TCGA-Omics-Datenanalyse gewonnen. Abbildungen 1A-1B zeigen die Heatmaps und Vulkanplots der 20 am stärksten hochregulierten/herunterregulierten unterschiedlich exprimierten mRNAs, während Abbildungen 1C-1D die Heatmaps und Vulkanplots der 20 am stärksten hochregulierten/herunterregulierten unterschiedlich exprimierten miRNAs zeigen. Um prognostische miRNAs auszuwählen, wurden die unterschiedlich exprimierten miRNAs mittels univariater Cox-Analyse bewertet. In der univariaten Cox-Analyse wurden 26 wichtige miRNAs mit signifikanten Veränderungen identifiziert, darunter sieben unterschiedlich exprimierte miRNAs (hsa-miR-499a-5p, hsa-miR-99a-5p, hsa-miR-337-3p, hsa-miR-4746-5p, hsa-miR-432-5p, hsa-miR-142-3p, hsa-miR-137-3p), die als unabhängige prognostische Faktoren für HNSCC-Patienten identifiziert wurden. Daher lautet die Formel für das Modell in dieser Studie: Risikoscore = (0,238 × hsa-miR-337-3p) + (0,246 × hsa-miR-99a-5p) - (0,232 × hsa-miR-4746-5p) - (0,233 × hsa-miR-432-5p) - (0,261 × hsa-miR-142-3p). Darüber hinaus wurden die Risikoscores für jeden Patienten in der Studienkohorte berechnet. Anschließend wurde der mediane Risikoscore als Schwellenwert verwendet, um die Patienten in Hochrisiko- und Niedrigrisikogruppen im Trainingssatz, Testdatensatz und bei allen Patienten zu unterteilen.
 Abb. 1. Das Vulkan-Diagramm und das Heatmap der 20 am stärksten hochregulierten/niedrigregulierten unterschiedlich exprimierten Gene oder miRNAs. A-B: mRNA; C-D: miRNAs.
Abb. 1. Das Vulkan-Diagramm und das Heatmap der 20 am stärksten hochregulierten/niedrigregulierten unterschiedlich exprimierten Gene oder miRNAs. A-B: mRNA; C-D: miRNAs.
3. Überlebens Ergebnisse und multivariate Analyse
In dieser Studie wurde der Ausdruck von sieben miRNAs in Bezug auf das Überleben der Patienten mithilfe von KM-Überlebenskurven analysiert. Es wurde festgestellt, dass hsa-miR-499a-5p, hsa-miR-99a-5p, hsa-miR-337-3p, hsa-miR-4746-5p, hsa-miR-432-5p, hsa-miR-142-3p und hsa-miR-137-3p signifikanten Einfluss auf die Gesamtüberlebensraten (OS) hatten (Abbildungen 2A-G). Darüber hinaus wurden die KM-Überlebenskurven für die beiden Datensätze verwendet, um den prädiktiven Wert der sieben miRNAs zu bewerten. In sowohl dem Trainingsset als auch dem Testset hatten Patienten in der Hochrisikogruppe niedrigere Überlebensraten als diejenigen in der Niedrigrisikogruppe. Eine ROC-Kurvenanalyse wurde durchgeführt, um zu bestimmen, ob die Ausdrucksmuster der überlebensassoziierten miRNAs eine frühe Vorhersage des Auftretens von HNSCC ermöglichen könnten. Die Ergebnisse zeigten eine AUC von 0,716 für das Trainingsset und eine AUC von 0,654 für das Testset, was auf eine moderate Sensitivität und Spezifität des prognostischen Modells hinweist. Die Risikolebensstatusgrafik zeigte, dass mit steigendem Risikoscore des Patienten auch die Sterberate zunahm (Abbildung 3). Um ein prognostisches Modell basierend auf den sieben miRNAs zu erstellen, wurden univariate und multivariate Cox-Analysen durchgeführt, um Risikofaktoren zu identifizieren. Die Ergebnisse zeigten, dass basierend auf den sieben miRNA-Eigenschaften Geschlecht und N-Stadium unabhängige prognostische Faktoren für das OS waren (Abbildung 4).
 Abb. 2. Gesamtüberlebensanalyse von 7 miRNA, dem Trainingssatz und dem Testsatz. A-G: miRNAs; H: Trainingssatz; I: Testsatz.
Abb. 2. Gesamtüberlebensanalyse von 7 miRNA, dem Trainingssatz und dem Testsatz. A-G: miRNAs; H: Trainingssatz; I: Testsatz.
 Abb. 3. Die Ergebnisse der ROC-Kurven und der Überlebensstatus der Patienten im Trainings- und Testdatensatz. A: ROC im Trainingsdatensatz; B: ROC im Testdatensatz; C: Überlebensstatus im Trainingsdatensatz; D: Überlebensstatus im Testdatensatz.
Abb. 3. Die Ergebnisse der ROC-Kurven und der Überlebensstatus der Patienten im Trainings- und Testdatensatz. A: ROC im Trainingsdatensatz; B: ROC im Testdatensatz; C: Überlebensstatus im Trainingsdatensatz; D: Überlebensstatus im Testdatensatz.
 Abb. 4. Univariate und multivariate COX-Analyse zur Identifizierung der Risikofaktoren. A: Univariat; B: Multivariat.
Abb. 4. Univariate und multivariate COX-Analyse zur Identifizierung der Risikofaktoren. A: Univariat; B: Multivariat.
4. miRNA-Zielgenanalyse
Die Vorhersage der Zielgene für diese sieben miRNAs ergab 60 Zielgene, die aus Online-Datenbanken gewonnen wurden. Die potenziellen Verbindungen zwischen miRNAs und Zielgenen wurden mit Cytoscape untersucht. Wie in Abbildung 5 gezeigt, war hsa-miR-137-3p der größte Knoten im Netzwerk, während hsa-miR-99a-5p und hsa-miR-4746-5p keine passenden Zielgene hatten (rote Knoten repräsentieren hochregulierte, grüne Knoten repräsentieren herunterregulierte). Um die biologischen Funktionen dieser Zielgene zu bewerten, wurden GO-Anreicherungs- und KEGG-Pfadanalysen durchgeführt. Die GO-Analyse ergab eine Anreicherung der Zielgene in biologischen Prozessen (BP) wie der negativen Regulation von zellulären Prozessen, der Gewebeentwicklung und der Reaktion auf hemmende Faktoren. Molekulare Funktionen (MF) waren angereichert in strukturellen Bestandteilen der extrazellulären Matrix, Wachstumsfaktorbindung, Aktinbindung und der Aktivität von Protein-Tyrosin/Serin/Threonin-Phosphatasen. Zelluläre Komponenten (CC) waren hauptsächlich in Thrombozyten-alpha-Granula, kondensierten Chromosomen-Kinetochoren, der extrazellulären Matrix und anderen Begriffen angereichert. Die KEGG-Pfadanalyse zeigte, dass diese Zielgene hauptsächlich in den Pentosephosphatweg, den Galaktosemetabolismus und die Glykolyse/Gluconeogenese-Pfade angereichert waren (Abbildung 6).
 Abb. 5. Cytoscape untersuchte die potenzielle Verbindung zwischen MikroRNA und Zielgenen.
Abb. 5. Cytoscape untersuchte die potenzielle Verbindung zwischen MikroRNA und Zielgenen.
 Abb. 6. Die Ergebnisse der GO-Analyse und der KEGG-Analyse der Zielgene.
Abb. 6. Die Ergebnisse der GO-Analyse und der KEGG-Analyse der Zielgene.
5. Beziehung zwischen miRNA
Zielgene und Überlebensausgang Die Analyse des Einflusses der miRNA-Zielgenexpression auf das Überleben der Patienten ergab, dass die Expression von neun Genen: CDCA4, CXCL14, FLNC, KLF7, NBEAL2, P4HA1, PFKM, PFN2 und SEPPINE1, das Gesamtüberleben (OS) signifikant beeinflusste (Abbildungen 7A-7I). Der Aufbau eines Protein-Protein-Interaktionsnetzwerks (PPI) offenbarte zwei zentrale Schlüsselgene, PDGFRB und RAD51 (Abbildung 7J).
 Abb. 7. Gesamtüberlebensanalyse der identifizierten Zielgene und des Protein-Protein-Interaktionsnetzwerks (PPI). A-I: mRNA; J: PPI-Netzwerk.
Abb. 7. Gesamtüberlebensanalyse der identifizierten Zielgene und des Protein-Protein-Interaktionsnetzwerks (PPI). A-I: mRNA; J: PPI-Netzwerk.
Fazit
Diese Studie, basierend auf dem TCGA-Datensatz, identifizierte ein innovatives miRNA-Feature und analysierte die Prognose von HNSCC. Die Forschung liefert neuartige Erkenntnisse für die Entwicklung zuverlässiger Werkzeuge zur genauen Vorhersage von Behandlungsergebnissen bei Krebs. Zu den Einschränkungen dieser Studie gehören jedoch die fehlende Validierung in klinischen Proben und die relativ kleine Stichprobengröße, die die statistische Aussagekraft einschränken könnte. Die identifizierten molekularen Marker sollten in weiteren unabhängigen Studien und funktionalen Experimenten validiert werden.
Referenz:
- Wu Z H, Zhong Y, Zhou T, u. a.miRNA-Biomarker zur Vorhersage der Gesamtüberlebensraten bei Plattenepithelkarzinomen im Kopf-Hals-Bereich. Genomik, 2021, 113(1): 135-141.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Chaperon-vermittelte Autophagie steuert proteomische und transkriptomische Wege zur Aufrechterhaltung der Aktivität von Gliom-Stammzellen.
Zeitschrift: Krebsforschung
Jahr: 2022
Zirkuläre DNA-Tumorviren erzeugen zirkuläre RNAs.
Zeitschrift: Mitteilungen der Nationalen Akademie der Wissenschaften
Jahr: 2018
Wiederholte Immunisierung mit ATRA-haltigem liposomalem Adjuvans transdifferenziert Th17-Zellen zu einem Tr1-ähnlichen Phänotyp.
Zeitschrift: Zeitschrift für Autoimmunität
Jahr: 2024
Die Rolle der Histonvariante H2A.Z.1 bei Gedächtnis, Transkription und alternativer Spleißung wird durch Lysinmodifikationen vermittelt.
Zeitschrift: Neuropsychopharmakologie
Jahr: 2024
FAK-Verlust reduziert die ERK-Phosphorylierung, die durch BRAFV600E induziert wird, um die intestinale Stammzelligkeit und die Bildung von Blinddarmtumoren zu fördern.
Journal: Elife
Jahr: 2023
Identifizierung von zirkulären RNAs, die die Proliferation von Kardiomyozyten in neonatalen Schweineherzen regulieren
Journal: JCI Insight
Jahr: 2024
Mehr ansehen Artikel, die von unseren Kunden veröffentlicht wurden.


