
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
oxBS-seq
DNA-Methylierung und Hydroxymethylierung sind wichtige epigenetische Modifikationen zur Regulierung der Genexpression. DNA-Methylierung unterdrückt typischerweise die Gentranskription. Hydroxymethylierung hingegen aktiviert die Genexpression oder fördert die DNA-Demethylierung.
5-Hydroxymethylcytosin (5hmC) wird aus 5-Methylcytosin (5mC) durch eine Gruppe von Enzymen, die als Ten-Eleven-Translokation (TET) Familie Dioxygenasen bezeichnet werden, umgewandelt. Die Goldstandard-Bisulfit-Konversionstechnologien zur Untersuchung der DNA-Methylierung unterscheiden nicht zwischen 5mC und 5hmC. Neue Ansätze zur kartografischen Erfassung von 5hmC im gesamten Genom haben sich jedoch schnell weiterentwickelt, obwohl unklar ist, wie die verschiedenen Methoden im Hinblick auf die genaue Identifizierung von 5hmC abschneiden.
CD Genomics kann drei Ansätze zur genome-weiten Erkennung von 5hmC anbieten:
- Ganzgenom-Bisulfid-/oxidative Bisulfid-SequenzierungWGBS/WGoxBS-seq)
- Reduzierte repräsentative Bisulfid-oxidative Bisulfid-SequenzierungRRBS/RRoxBS)
- Antikörperbasierte Immunpräzipitation und Sequenzierung von Hydroxymethylierter DNAhMeDIP-seq).
- Zielgerichtete DNA-Bisulfid-/Hydroxymethylierung-SequenzierungTBS/ oxTBS-seq)
- 5hmC-selektive chemische Markierungstechnologie (5hmC-Seal)
Die Einführung von oxBS-seq
5-Methylcytosin (5mC) kann von einer Gruppe von Oxygenasen, den Ten-Eleven-Translokationsenzymen (Tets), oxidiert werden. Unter Verbrauch von Sauerstoff und 2-Oxoglutarat oxidieren diese Fe(II)-abhängigen Dioxygenasen 5mC in einer ersten Reaktion zu 5-Hydroxymethylcytosin (5hmC), gefolgt von 5-Formylcytosin (5fC) und schließlich 5-Carboxycytosin (5caC). Die häufigste Form dieser oxidierten Cytosinvarianten ist 5hmC.
Die oxidative Bisulfite-Konversion (oxBS), eine Vor-Bisulfite-Oxidationsreaktion, wandelt 5hmC in 5fC um, das durch Bisulfite in 5f-Uracil und in Thymidin in der nachfolgenden PCR umgewandelt wird. Diese Bibliothek wird dann mit einer traditionellen Bisulfite-Sequenzierungsbibliothek (BS-seq), die aus derselben Probe erstellt wurde, verglichen, um die Menge an 5mC und 5hmC für jedes modifizierte Cytosin innerhalb der DNA zu bestimmen. oxBS-seq hat einen relativ einfachen und schnellen experimentellen Arbeitsablauf und kann sowohl das Methylom als auch das Hydroxymethylom gleichzeitig erfassen.
Wenn Sie mehr über die Einführung von oxBS-seq erfahren möchten, können Sie unseren Artikel lesen: "oxBS-Seq, eine epigenetische Sequenzierungsmethode zur Unterscheidung von 5mC und 5hmC".
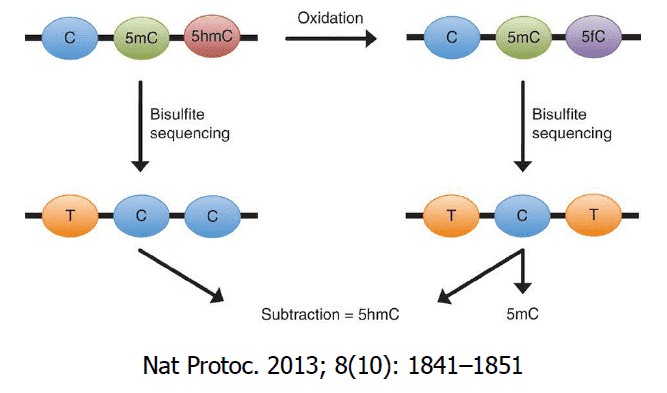 Abbildung 1. Schematische Darstellung des oxBS-seq-Prinzips. (Booth et al., 2013)
Abbildung 1. Schematische Darstellung des oxBS-seq-Prinzips. (Booth et al., 2013)
Was sind die Vorteile von oxBS-seq?
- Die Auflösung auf Einzel-Nukleotid-Ebene umfasst sowohl CpG- als auch non-CpG-Methylierung im gesamten Genom.
- Die Dichte von 5mC in repetitiven Regionen und Regionen mit niedrigerer Dichte ist abgedeckt.
- Die Methoden unterscheiden klar zwischen 5mC und 5hmC, was eine präzise Identifizierung von 5mC ermöglicht.
- Etablierung eines neuartigen "Goldstandards" für die DNA-Methylierungsdetektion
- Genomweite Einzel-Nukleotid-Erkennung von DNA-Hydroxymethylierungsmodifikationen
- Multi-Kriterien-Validierung für hohe Oxidationseffizienz und hohe Bisulfit-Umwandlungsrate
- Der experimentelle Bias ist minimal, mit hoher Reproduzierbarkeit (R²>0,98).
- Zufriedenstellung vielfältiger Anforderungen an Sequenzierungsanwendungen: Optimierte genomweite oxidationsmethylierungssequenzierung (oxRRBS), gezielte Regionen-Oxidationsmethylierungssequenzierung (Target-oxBS)
Was sind die Anwendungen von oxBS-seq?
Unsere oxBS-seq-Analyse bietet vielseitige Anwendungen in verschiedenen Forschungsbereichen:
- Epigenetik: Unsere oxBS-seq-Analyse ermöglicht eine differenzierte Untersuchung des Zusammenspiels zwischen DNA-Methylierung und Hydroxymethylierung in verschiedenen biologischen Szenarien und bietet ein nuanciertes Verständnis der epigenetischen Modifikationen, die mit der Genregulation, zellulären Differenzierung und Entwicklungswegen verwoben sind.
- Krankheitsforschung: Durch sorgfältige Untersuchung der DNA-Methylierungs- und Hydroxymethylierungsmuster in Krankheitsproben enthüllt unsere oxBS-seq-Analyse epigenetische Veränderungen, die mit Erkrankungen wie Krebs, neurologischen Störungen und Herz-Kreislauf-Erkrankungen verbunden sind.
- Umwelt- EpigenomikWeit verbreitet zur Untersuchung des Einflusses von Umwelteinflüssen auf die Dynamik der DNA-Methylierung und Hydroxymethylierung eingesetzt, unterstützt unsere oxBS-seq-Analyse dabei, epigenetische Veränderungen zu erkennen, die durch unterschiedliche Umweltbelastungen induziert werden, und beleuchtet somit deren potenzielle Auswirkungen auf Gesundheit und Krankheitsanfälligkeit.
- Entwicklungsbiologie: Durch die Erleichterung der Erforschung der Dynamik der DNA-Methylierung während der embryonalen Entwicklung und der Gewebedifferenzierungsprozesse liefert unsere oxBS-seq-Analyse komplexe Methylierungslandschaften in hoher Auflösung.
oxBS-seq Sequenzierungs-Workflow
Genomisches DNA wurde verarbeitet, um Fragmente von 10 kb zu erhalten. Fragmentierte DNA wurde mithilfe von Reinigungssäulen konzentriert. Diese fragmentierte und gereinigte DNA wurde für die Oxidation + Bisulfidbehandlung verwendet. Pufferwechsel, Denaturierung, Oxidation, Bisulfidumwandlung, Reinigung und qualitative Bewertung der Oxidation von 5-Hydroxymethylcytosin wurden durchgeführt. Schließlich wurden oxBS- und BS-konvertierte DNA verwendet, um oxBS-seq- und entsprechende BS-seq-Bibliotheken zu erstellen. Alle doppel- und einzelsträngigen DNA wurden vor der Clustererzeugung und Sequenzierung auf der Illumina-Plattform gemäß den Herstellervorgaben auf einem Agilent 2100 Bioanalyzer quantifiziert.
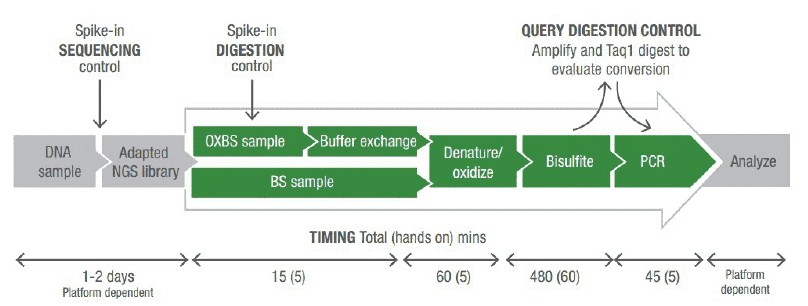 Abbildung 2. Schematische Darstellung der Schritte für das Hochdurchsatz-Sequenzieren des Ig-Sequenzrepertoires (Georgiou et al., 2014).
Abbildung 2. Schematische Darstellung der Schritte für das Hochdurchsatz-Sequenzieren des Ig-Sequenzrepertoires (Georgiou et al., 2014).
Dienstspezifikation
Musteranforderungen
|
|
 |
Sequenzierung
|
 |
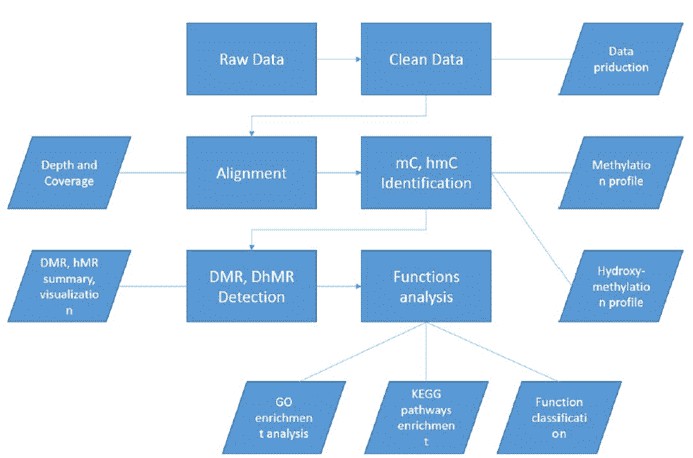
Bioinformatikanalyse Wir bieten maßgeschneiderte bioinformatische Analysen an, einschließlich:
|
Analyse-Pipeline
Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details in oxBS-seq für Ihr Schreiben (Anpassung)
Referenzen:
- Giehr, Pascal, Kyriakopoulos, et al. Zwei sind besser als einer: HPoxBS - Haarpin-oxidative Bisulfite-Sequenzierung. Nukleinsäurenforschung, 2018.
- Skvortsova K, Zotenko E, Luu P L, et al. Umfassende Bewertung von genomweiten Ansätzen zur Profilierung von 5-Hydroxymethylcytosin in menschlicher DNA. Epigenetik & Chromatin, 2017, 10(1):16.
- Booth M J, Ost T W B, Beraldi D, et al. Oxidative Bisulfit-Sequenzierung von 5-Methylcytosin und 5-Hydroxymethylcytosin. Naturprotokolle, 2013, 8(10): 1841-1851.
- Han Y, Ji L, Guan Y, et al. Eine epigenomische Landschaft der zervikalen intraepithelialen Neoplasie und des Gebärmutterhalskrebses unter Verwendung von Methylom und Hydroxymethylom mit Einzelbasenauflösung. Klinische und translationale Medizin, 2021, 11(7): e498.
Demo-Ergebnisse
 (Han et al., 2021)
(Han et al., 2021)
oxBS-seq häufig gestellte Fragen (FAQs)
Was ist oxBS-seq?
oxBS-seq ist ein Hochdurchsatz-Sequenzierung Technik zur Messung der Verteilung von DNA-Methylierung und Hydroxymethylierung. Sie kombiniert oxidative Bisulfitbehandlung (oxBS) mit Sequenzanalyse, um 5mC und 5hmC mit einer Einzelbasenauflösung zu unterscheiden und detaillierte Informationen über diese beiden Modifikationen im Genom bereitzustellen.
2. Wie unterscheidet sich oxBS-seq von traditionellem BS-seq?
Traditionelles BS-seq (Bisulfid-Sequenzierung) wandelt unmethylierte Cytosine (Cs) in Uracile (Us) um, während methylierte Cs erhalten bleiben. Allerdings wird 5hmC ebenfalls durch BS in T umgewandelt, wodurch es von 5mC nicht zu unterscheiden ist. Im Gegensatz dazu oxidiert oxBS-seq chemisch 5hmC zu C vor der BS-Behandlung, was eine Unterscheidung zwischen 5mC und 5hmC ermöglicht.
3. Wie ist der Workflow von oxBS-seq?
Der oxBS-seq-Workflow umfasst mehrere wichtige Schritte: DNA-Extraktion, oxidative Methylierung, Bisulfidbehandlung (BS), Sequenzierung und anschließende Datenanalyse. Zunächst wird 5hmC zu C oxidiert, gefolgt von der BS-Behandlung, um unmethylierte Cs in Ts umzuwandeln, während 5mC und 5hmC erhalten bleiben. Anschließend werden Sequenzierungsdaten generiert und einer umfassenden Analyse unter Verwendung bioinformatischer Werkzeuge unterzogen.
4. Welche Analysemethoden gibt es für oxBS-seq-Daten?
Die angewandten analytischen Rubriken auf oxBS-seq-Daten umfassen mehrere Aspekte: Vorverarbeitung, Ausrichtung, Bewertung der Methylierungsniveaus und Analyse der differentiellen Modifikationen. Der Schritt der Vorverarbeitung umfasst die Aufgaben der Qualitätskontrolle und der Sequenzkürzung. Die Ausrichtung ist eine Aufgabe, die das Abgleichen der Sequenzierungsdaten mit einem Referenzgenom erleichtert, wodurch die Schätzung der Methylierungsniveaus an jedem Standort ermöglicht wird und eine Unterscheidung zwischen 5mC und 5hmC erlaubt wird. Eine Reihe nachfolgender statistischer Analysen und biologischer Interpretationen wird eingesetzt, um Schlussfolgerungen zu ermitteln, die für die zentrale Forschungsfrage relevant sind.
5. Wie entwirft man oxBS-seq-Experimente?
Die Planung von oxBS-seq-Experimenten erfordert die Durchführung bestimmter Aufgaben wie die Auswahl der Proben, die Ausarbeitung des experimentellen Designs, die Bestimmung der Sequenzierungstiefe und die Strategie für die Datenanalyse. Um eine Vielzahl von Forschungsfragen ganzheitlich zu adressieren, kann der Experimentator die Experimentparameter optimieren, um die Zuverlässigkeit und Genauigkeit der experimentellen Ergebnisse zu erhöhen. Darüber hinaus ist es wichtig, dass wirksame experimentelle Kontrollen und Replikate einbezogen werden, um die Reproduzierbarkeit und Stabilität der Ergebnisse zu gewährleisten.
6. Was sind die Einschränkungen von oxBS-seq?
Obwohl oxBS-seq mit zahlreichen Vorteilen aufwarten kann, ist es nicht ohne Einschränkungen. Beispielsweise können ein Spektrum niedriger Methylierungsgrade oder Bereiche mit geringer genomischer Komplexität potenziell die Genauigkeit der Technik beeinträchtigen. Darüber hinaus kann der technische Aufwand, der von den experimentellen Verfahren und der anschließenden Datenanalyse gefordert wird, erhebliche Herausforderungen mit sich bringen.
oxBS-seq Fallstudien
Genomweite DNA-Methylierungs- und Hydroxymethylierungsänderungen zeigten die epigenetische Regulation von Neuromodulation und Myelinisierung im Hypothalamus von Yaks.
Zeitschrift: Frontiers in Genetics
Impact Faktor: 4,772
Veröffentlicht: 27. September 2021
Hintergrund
5-Methylcytosin (5mC) und 5-Hydroxymethylcytosin (5hmC) sind bedeutende epigenetische Modifikationen, die integraler Bestandteil des Neuroentwicklungsprozesses sind. Es gibt jedoch nur wenige Forschungen, die genomweite Muster von 5mC und 5hmC in Gehirnregionen von Tieren als Funktion natürlicher Hochgebirgsumgebungen identifizieren.
Methoden
- Yak und Rinder
- RNA- und DNA-Extraktion
- Reduzierte Repräsentation Bisulfid-Sequenzierung
- Oxidative reduzierte Repräsentations-Bisulfid-Sequenzierung
- Identifizierung von unterschiedlich methylierten Regionen
- Identifikation von unterschiedlich Hydroxymethylierten Regionen
Ergebnisse
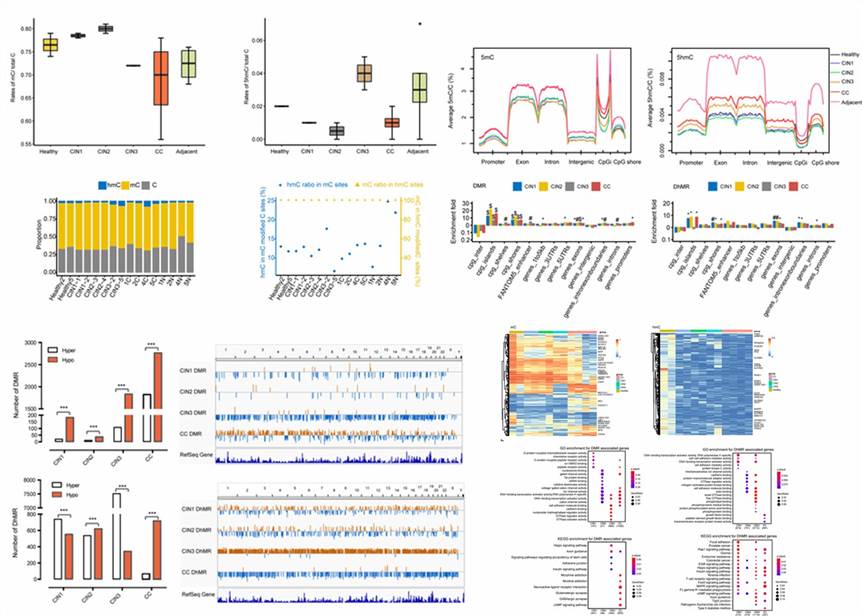
RRBS und oxRRBS bieten eine Einzelbasenauflösung zur Profilierung von Methylierung und Hydroxymethylierung über 3,28 Millionen CpG-Stellen, wobei oxRRBS eine höhere Korrelation zwischen den komplementären Strängen zeigt. Die geringere Konsistenz in RRBS könnte auf die dynamische Beteiligung von 5hmC an der DNA-Demethylierung zurückzuführen sein. Die Analyse, die sich auf "echte" 5hmC-Stellen konzentrierte, zeigte gewebespezifische Unterschiede zwischen Yak und Rind, insbesondere im Hypothalamus, Kleinhirn und Hirnstamm.
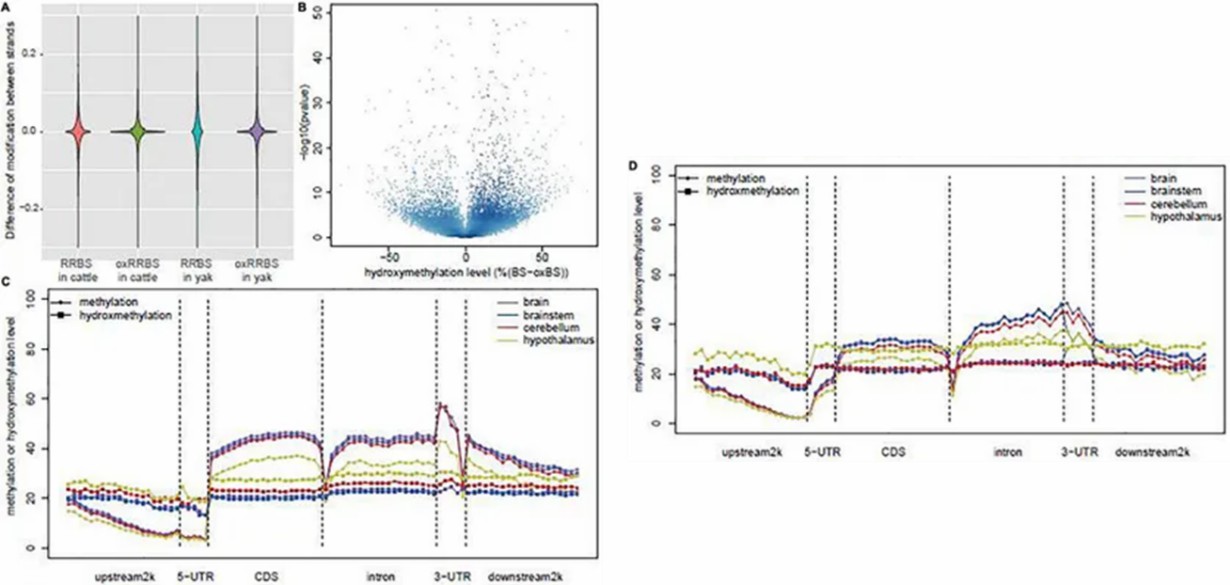 Abb. 1. Globale DNA-Methylierung und Hydroxymethylierung zwischen den Proben.
Abb. 1. Globale DNA-Methylierung und Hydroxymethylierung zwischen den Proben.
Die Studie erläuterte unterschiedliche Verteilungsmuster der 5mC- und 5hmC-Spiegel über Gene hinweg und hob signifikante Unterschiede zwischen den Geweben hervor, insbesondere im Hypothalamus. Paarweise Vergleiche zwischen den Gehirnen von Yak und Rind zeigten unterschiedliche Methylierungs- und Hydroxymethylierungsregionen (DMRs und DhMRs), die hauptsächlich durch veränderte 5mC- und 5hmC-Spiegel bedingt waren. Die Anreicherungsanalyse der Gene-Ontologie (GO)-Begriffe unterstrich gewebespezifische regulatorische Mechanismen, insbesondere in nervensystembezogenen Signalwegen, was auf eine komplexe epigenetische Regulation der Gehirnfunktion über Arten hinweg hindeutet.
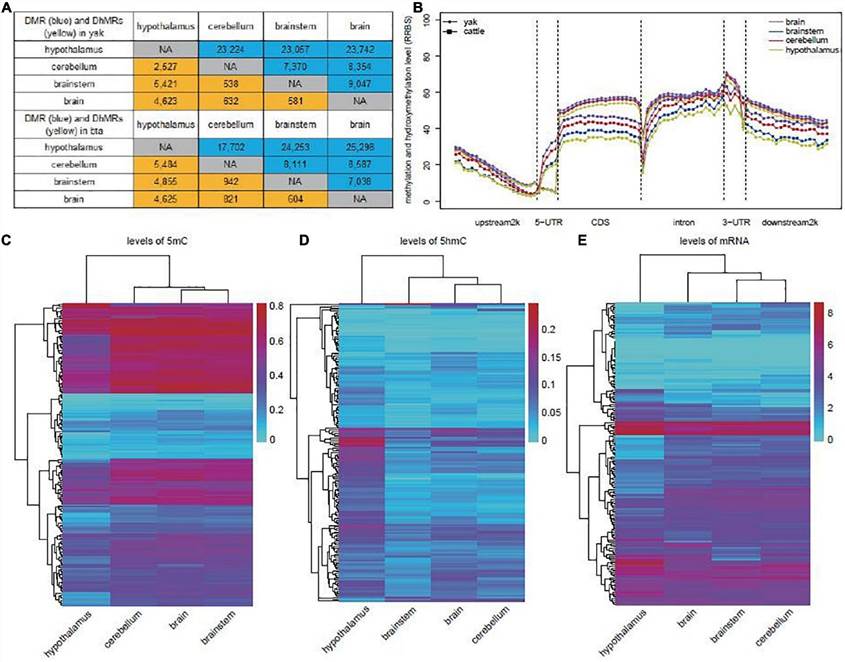 Abb. 2. Identifizierung von DMRs und DhMRs bei Yak und Rind.
Abb. 2. Identifizierung von DMRs und DhMRs bei Yak und Rind.
Fazit
Diese Studie nutzt RRBS- und oxidative RRBS (oxRRBS)-Techniken, um signifikante Unterschiede in 5mC und 5hmC im Hypothalamus und anderen Gehirnregionen von Yaks und Kühen aufzudecken. Dazu gehört die Identifizierung von unterschiedlich methylierten Regionen (DMRs) und unterschiedlich hydroxymethylierten Regionen (DhMRs), von denen die meisten überlappen. Folglich wurden unterschiedlich exprimierte Gene (DEGs), die potenziell durch DMRs und DhMRs reguliert werden und möglicherweise eine entscheidende Rolle bei der neuronalen Regulation und der Myelinbildung spielen, validiert. Zusammenfassend deuten die Ergebnisse darauf hin, dass die epigenetische Regulation, die durch 5mC und 5hmC vermittelt wird, erheblichen Einfluss auf die Entwicklung und biologischen Funktionen des Hypothalamus haben könnte, was möglicherweise zur Verbesserung der physiologischen Anpassungsfähigkeit unter Hochgebirgsbedingungen beiträgt.
Referenz:
- Chai Z, Wu Z, Ji Q, et al. Genomweite Veränderungen der DNA-Methylierung und Hydroxymethylierung zeigten die epigenetische Regulation der Neuromodulation und Myelinisierung im Hypothalamus von Yaks. Grenzen der Genetik, 2021, 12: 592135.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Die Spaltung von Phagen-DNA durch Restriktionsendonukleasen ermöglicht die Wiederbelebung aus der durch Cas13 induzierten bakteriellen Dormanz.
Zeitschrift: Nature Mikrobiologie
Jahr: 2023
IL-4 fördert die Erschöpfung von CD8.+ CART-Zellen
Zeitschrift: Nature Communications
Jahr: 2024
Fettreiche Diäten während der Schwangerschaft führen zu Veränderungen der DNA-Methylierung und Proteinexpression im Pankreasgewebe der Nachkommen: Ein Multi-Omics-Ansatz
Internationale Zeitschrift für Molekulare Wissenschaften
Jahr: 2024
KMT2A assoziiert mit dem PHF5A-PHF14-HMG20A-RAI1 Subkomplex in Stammzellen des Pankreaskarzinoms und reguliert epigenetisch deren Eigenschaften.
Zeitschrift: Nature Communications
Jahr: 2023
Krebsassoziierte DNA-Hypermethylierung von Polycomb-Zielen erfordert die duale Erkennung von Histon H2AK119-Ubiquitinierung und der sauren Tasche des Nukleosoms durch DNMT3A.
Journal: Wissenschaftliche Fortschritte
Jahr: 2024
Genomisches Imprinting-ähnliches monoalleles väterliches Ausdrucksmuster bestimmt das Geschlecht von Kanalkatzenfischen.
Journal: Wissenschaftliche Fortschritte
Jahr: 2022
Mehr ansehen Artikel, die von unseren Kunden veröffentlicht wurden.


