
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
CD Genomics bietet seit einigen Jahren einen flexiblen und erschwinglichen Whole Exome Sequencing-Service an. Wir verwenden die Illumina HiSeq-Sequenzierungsplattform, um die Informationen über genetische Variationen effizienter zu erhalten.
WGS vs WES vs gezielte Panels: Ein schneller Leitfaden zur Methodenauswahl
Nicht sicher, ob Sie Breite, Tiefe oder eine fokussierte Genliste benötigen? Der falsche Test kann zu zu vielen Daten, nicht genügend Tiefe oder fehlenden wichtigen Variantentypen führen. Verwenden Sie den Fluss unten, um den Umfang mit Ihrer Forschungsfrage abzugleichen.
Schneller Entscheidungsfluss
Q1. Benötigen Sie eine genomweite Entdeckung über kodierende Regionen hinaus, oder liegt der Schwerpunkt auf struktureller Variation?
- Ja WGS
- Nein → Gehe zu Q2
Q2. Ist Ihr Ziel eine umfassende Entdeckung oder das Screening von kodierenden Varianten über viele Gene hinweg?
- Ja WES
- Nein → Gehe zu Q3
Q3. Haben Sie bereits eine definierte Genliste / enge Hypothese?
- Ja Zielgerichtetes Panel
- Nein WES
Q4. Benötigen Sie eine sehr hohe Tiefe bei einer begrenzten Anzahl von Loci?
- Ja Zielgerichtetes Panel (oder benutzerdefinierte Zielgruppen)
- Nein WES
Empfohlene Strategie
- Beste Balance für die Entdeckung von Codierungsvarianten bei überschaubarem Datenvolumen.
- Ideal, wenn Sie Entdeckungen jenseits einer festen Genliste benötigen, ohne die Komplexität des gesamten Genoms.
- Am besten für maximale Breite und Projekte, die genomweite Signale benötigen.
- Erwarten Sie ein höheres Datenvolumen und eine komplexere Analyse der nachgelagerten Daten.
Gezielte Genpanel-Sequenzierung
- Am besten für eine feste Genliste und hochauflösende, zielgerichtete Lesungen.
- Nicht für die Entdeckung außerhalb des Panel-Designs vorgesehen.
1-Minuten-Vergleichstabelle
| Entscheidungsfaktor | WGS | WES | Zielgerichtetes Panel |
|---|---|---|---|
| Umfang | genomweit | Protein-kodierendes Exom | Ausgewählte Gene/Regionen |
| Am besten für | Maximale Breite | Codierungsentdeckung | Hypothesenbasierte Ziele |
| Datenskala | Hoch | Mittel | Niedrig |
| Tiefenstrategie | breit | Ausgewogen | Sehr hoch bei den Zielen |
| Interpretationsaufwand | Höher | Moderat | Niedriger |
| Grenzen | Mehr Datenverarbeitung | Begrenztes äußeres Exom | Begrenztes Außenpaneel |
Mehr erfahren
- Leitfaden: Whole Exom vs. Whole Genome Sequenzierung
Die Einführung der gesamten Exom-Sequenzierung
Das menschliche Genom umfasst ungefähr 3×10.9 Basen und enthält ungefähr 180.000 kodierende Regionen (Exom), die etwa 1,7 % des menschlichen Genoms ausmachen. Es wird geschätzt, dass 85 % der krankheitsverursachenden Mutationen im Exom auftreten. Aus diesem Grund hat die Sequenzierung des gesamten Exoms das Potenzial, eine höhere Ausbeute an relevanten Varianten zu einem viel niedrigeren Preis zu entdecken als Whole-Genome-SequenzierungDie gesamte Exomsequenzierung wird als eine effiziente und leistungsstarke Methode angesehen, um die genetischen Varianten zu identifizieren, die erblich bedingte Phänotypen beeinflussen, einschließlich wichtiger krankheitsverursachender Mutationen und natürlicher Variationen, die zur Verbesserung von Pflanzen und Vieh eingesetzt werden können.
Die gesamte Exom-Sequenzierung nutzt die Exom-Erfassungstechnologie, um Exons anzureichern, und sequenziert diese Regionen dann in einem Hochdurchsatzverfahren. Genauer gesagt werden DNA-Proben zunächst fragmentiert, und biotinylierte Oligonukleotid-Sonden (Baits) werden verwendet, um selektiv an das Exom im Genom zu hybridisieren. Magnetische Streptavidin-Perlen werden dann verwendet, um an die biotinylisierten Sonden zu binden. Der nicht zielgerichtete Teil des Genoms wird abgewaschen, und die PCR wird verwendet, um die Probe für DNA aus dem Zielbereich anzureichern. Anschließend wird die Probe mit der Illumina HiSeq-Plattform sequenziert. Diese Strategie kann zu einer bis zu 100-fachen Verbesserung der Genabdeckung für das menschliche Genom führen. Die validierten Sequenzdaten werden dann für die Variantenanalyse und Forschungsinterpretation verwendet.
Unsere Exom-Sequenzierungslösungen
Bei CD Genomics bieten wir maßgeschneiderte Exom-Sequenzierung Dienstleistungen für Mensch/Maus und Tier/Pflanze Genome, die präzise Variantenbestimmung und kosteneffiziente Lösungen bieten.
| Diensttyp | Empfohlene Datengröße | Notizen |
|---|---|---|
| Human/Maus Exom-Sequenzierung | ||
| - Kernpanel | ≥8 Gb @ 100X | Optimiert für Abdeckung und Effizienz |
| - Vererbtes Panel | ≥11 Gb @ 100X | Erweiterte SNV/InDel/CNV-Erkennung |
| - Tumorpanel | ≥20 Gb @ 200X | Unterstützt TMB, MSI und Fusionsnachweis |
| Tier-/Pflanzen-Exom-Sequenzierung | Variiert je nach Art | Gezielte Sequenzierung für verschiedene Arten (z. B. Weizen, Mais, Rinder) |
Erkunden Sie unser Human/Maus Exom-Sequenzierungsdienste oder Tier-/Pflanzen-Exom-Sequenzierungsoptionen um die perfekte Lösung für Ihre Forschung zu finden. Diese prägnante Lösung hilft Ihnen, den geeigneten Exom-Sequenzierungsdienst für Ihre Forschungsbedürfnisse auszuwählen.
Vorteile der gesamten Exom-Sequenzierung
- Niedrigere Kosten und breite Verfügbarkeit
- Erhöhte Sequenzabdeckung (über 120X)
- Erkennung von kodierenden Einzel-Nukleotid-Polymorphismen (SNP) Varianten so empfindlich wie Whole-Genome-Sequenzierung
- Ein kleinerer Datensatz für schnellere und einfachere Analysen im Vergleich zur gesamten Genomsequenzierung.
- Medizinische und landwirtschaftliche Anwendungen
Workflow für die gesamte Exomsequenzierung
CD Genomics nutzt das Illumina HiSeq-System, um schnelle und präzise Whole-Exome-Sequenzierung und bioinformatische Analysen anzubieten. Unser hochqualifiziertes Expertenteam führt das Qualitätsmanagement durch und folgt jedem Verfahren, um zuverlässige und unvoreingenommene Ergebnisse zu gewährleisten. Der allgemeine Arbeitsablauf für die Whole-Exome-Sequenzierung ist unten skizziert.

Beispielanforderungen
Die Projektanforderungen können je nach Art, Fangdesign und Studienzielen variieren. Wir werden die endgültigen Eingabe-/QC-Schwellenwerte während der Entwurfsprüfung bestätigen.
| Probenart | Empfohlene Eingabe (Richtlinie) | QC-Vorschläge | Notizen |
|---|---|---|---|
| Genomisches DNA (gDNA) | ≥ 500 ng | OD260/280 ~1,8–2,0; Qubit-Quantifizierung; minimale Degradation | Bevorzugt für standardisierte WES-Workflows |
| Blut / Zellen / Frisches Gewebe (als DNA-Quelle) | Wie erforderlich, um ≥ 500 ng gDNA zu extrahieren | Bitte geben Sie Informationen zur Extraktionsmethode an, falls verfügbar. | Wir können bei Bedarf zur Extraktion beraten. |
| FFPE-Gewebe (als DNA-Quelle) | Projektabhängig (oft höhere Eingaben) | DV200/Fragmentierungsbewertung empfohlen | Die Einheitlichkeit der Erfassung kann beeinträchtigt werden; planen Sie die Qualitätskontrolle konservativ. |
| Niedriginputproben | Projektabhängig | Vorausbewertung empfohlen | Fragen Sie uns nach Machbarkeit und Zielgruppenstrategie. |
Was wir überprüfen (typisch)DNA-Menge, Reinheit, Integrität/Fragmentierung (sofern zutreffend), Qualitätskontrollpunkte der Bibliothek (z. B. Größenverteilung, Komplexitätsindikatoren).
Sequenzierungsstrategie
1. Bibliothek & ErfassungHybridisierungsbasiertes Exom-Capturing (typische Zielgröße 35–65 Mb).
2. Sequenzierungsmodus: Paarend-End PE150 (150 bp × 2), dual-indizierte Bibliotheken.
3. Abdeckungsoptionen (durchschnittliche Zieltiefe):
- Standard WES: ~100×
- Deep WES: ~200×
- Hochauflösende Option: ~300× (projektabhängig)
4. Ungefähr Daten pro Probe (roh):
- 100×: ~8–12 Gb
- 200×: ~15–22 Gb
- 300×: ~22–30 Gb
5. QC-BerichterstattungZielgenaue Leistung, Duplikation und Verteilungsabdeckung (einschließlich ≥20× Abdeckungszusammenfassung über die Ziele).
Bioinformatische Analyse
Ein standardisiertes, reproduzierbares Bioinformatik Pipeline zur Umwandlung von Rohdaten in interpretierbare Varianten- und QC-Ausgaben.
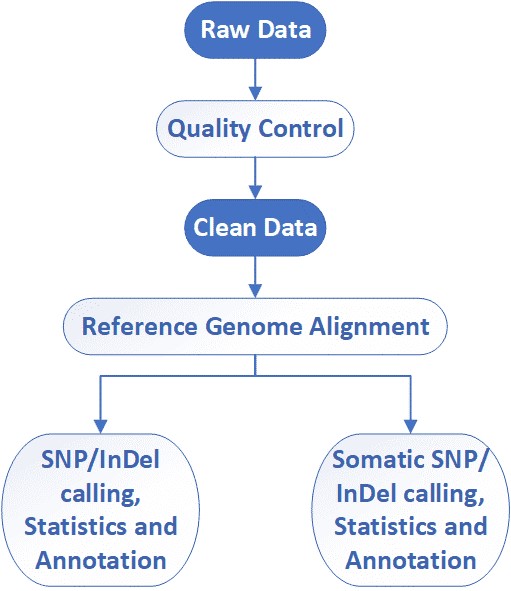
Kernpipeline (inklusive):
- Rohdaten-QC:
- Ausrichtung
- Abdeckung & Erfassung QC
- Variant-Erkennung
- Variantenannotation
- Zusammenfassende Berichterstattung
Optionale Zusatzleistungen (nach Bedarf auswählen):
- Kohortenebene gemeinsame Analyse
- CNV-Inferenz aus Exomdaten
- Familienbasierte Analyse
- Benutzerdefinierte Filterregeln und Priorisierung, die auf Ihren Hypothesen/Gensets basieren.
- Unterstützung bei der downstream-Interpretation
Was Sie erhalten werden (Liefergegenstände)
Daten Dateien (analysebereit):
- Rohdaten: FASTQ (+ QC-Zusammenfassungen)
- Ausrichtete Reads: BAM/CRAM (+ Ausrichtungsmetriken)
- Variantaufrufe: VCF für SNVs/Indels (und optionale CNV-Ausgaben, falls ausgewählt)
- Annotierte Variantentabellen: TSV/Excel-artige Tabellen mit konfigurierbaren Spalten für Filterung/Priorisierung
- QC- und Abdeckungs-Paket: Schlüsselkennzahlen zur Erfassung/Abdeckung, Zusammenfassungen zu Zielgenauigkeit und Uniformität, Duplikations- und Bibliotheksindikatoren
Berichte & Zusammenfassungen:
- Projektbericht: prägnante Übersicht über die Qualitätskontrolle der Proben, Sequenzierungs-/Erfassungsleistung und Gesamtergebnisse
- Variantenübersicht: Zählungen nach Typ, Verteilungssummen und Vergleichsübersichten auf Kohortenebene (wenn der Kohorten-Workflow ausgewählt ist)
Wiederverwendbare Ressourcen (für dein Team):
- Klare Dateibenennung + Ordnerstruktur für die Pipeline-Integration
- Methodenbereitstellung zur Beschreibung der Analyse Schritte für interne Dokumentation und Manuskriptvorbereitung (RUO)
CD Genomics bietet ein umfassendes Paket für die gesamte Exomsequenzierung an, das die Standardisierung von Proben, die Exom-Erfassung, den Bibliotheksaufbau, die Tiefensequenzierung, die Qualitätskontrolle der Rohdaten und die bioinformatische Analyse umfasst. Wir können diese Pipeline auf Ihre Forschungsinteressen zuschneiden. Wenn Sie zusätzliche Anforderungen oder Fragen haben, zögern Sie bitte nicht, uns zu kontaktieren.
Referenz:
- Warr A, Robert C, Hume D, et al. Exom-Sequenzierung: aktuelle und zukünftige Perspektiven. G3: Gene, Genom, Genetik, 2015, 5(8): 1543-1550.
Demonstrationsergebnisse
Diese Beispielgrafiken heben typische WES-Ausgaben hervor – Abdeckung/QC-Leistung, Variantenübersicht, Anreicherungsübersicht und genomweite Signalvisualisierung – für eine schnelle Projektüberprüfung.
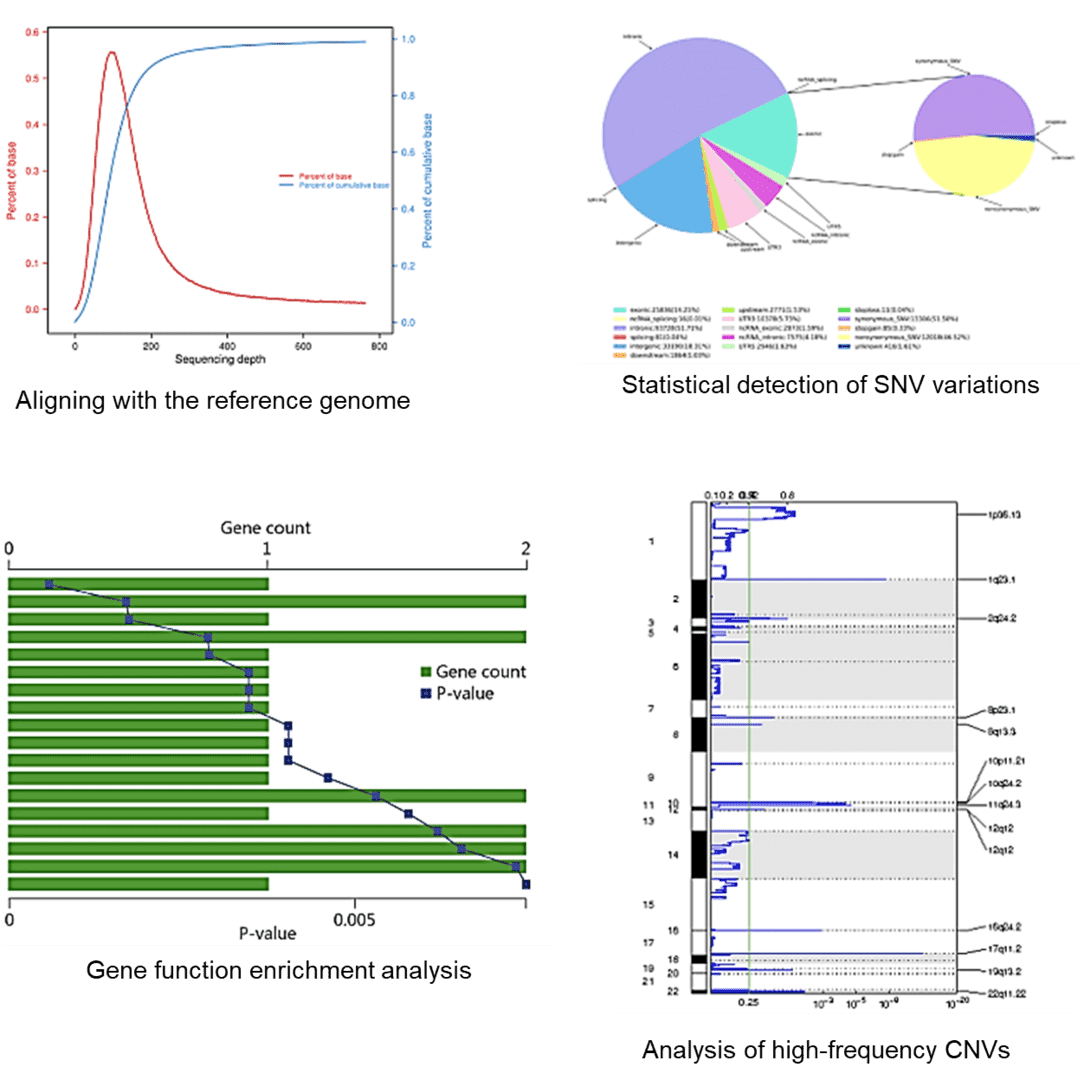
Häufig gestellte Fragen zur gesamten Exom-Sequenzierung
1. Was sind die Anwendungen der gesamten Exomsequenzierung?
Das menschliche Genom enthält ungefähr 180.000 kodierende Regionen (Exom), die etwa 1,7 % des menschlichen Genoms ausmachen. Es wird geschätzt, dass 85 % der krankheitsverursachenden Mutationen im Exom auftreten. Daher ist die gesamte Exomsequenzierung ein potenzieller Beitrag zum Verständnis menschlicher Krankheiten. Die gesamte Exomsequenzierung ist ein kosteneffektives und leistungsstarkes Werkzeug, das besonders für größere Stichprobengrößen und hohe Abdeckung geeignet ist. Die gesamte Exomsequenzierung wird hauptsächlich verwendet, um die genetischen Ursachen sowohl von Mendelschen als auch von häufigen Krankheiten wie Krebs und Diabetes zu untersuchen.
 Abbildung 1. Anwendung der gesamten Exomsequenzierung bei einer komplexen Erkrankung.
Abbildung 1. Anwendung der gesamten Exomsequenzierung bei einer komplexen Erkrankung.
2. Welche Variationen kann die gesamte Exomsequenzierung erkennen?
Die gesamte Exom-Sequenzierung kann SNPs, InDels und möglicherweise Kopienzahlvariationen (CNVs) nachweisen.
3. Wie bestimme ich die Sequenzierungstiefe?
Die Sequenzierungstiefe ist ein wichtiger Faktor für Hochdurchsatz-SequenzierungEine in der Zeitschrift Genomics & Informatics veröffentlichte Studie zeigte, dass die Sequenzierungstiefe des gesamten Exoms die Entdeckungsraten von Variationen beeinflussen kann. Zusammenfassend lässt sich sagen, dass die Anzahl der schädlichen SNPs und InDels, die in den kodierenden Regionen nachgewiesen wurden, bei Tiefen von mehr als 120× nur schwach anstieg. Mit anderen Worten, eine Sequenzierungstiefe von 120× kann als angemessen betrachtet werden, wenn die Exom-Capture-Sequenzierungstechnik verwendet wird, um signifikante Variationen in diagnostischen Studien zu identifizieren.
4. Was sind die Nachteile der gesamten Exomsequenzierung?
Die gesamte Exomsequenzierung zeichnet sich durch niedrigere Kosten, eine erhöhte Sequenzabdeckung sowie eine empfindliche und spezifische Identifizierung aus. Dennoch kann die gesamte Exomsequenzierung strukturelle Varianten nicht erkennen und bietet eine eingeschränkte Sicht, d.h. nur auf kodierende Regionen. Nicht alle Ziele werden erfasst (ungefähr 80%), und es ist schwierig, GC-reiche Regionen zu erfassen.
5. Was ist der Unterschied zwischen WES und WGS?
Die gesamte Exom-Sequenzierung (WES) zielt auf protein-codierende Regionen ab, um eine effiziente Variantenentdeckung bei überschaubarem Datenvolumen zu ermöglichen, während die gesamte Genom-Sequenzierung (WGS) das gesamte Genom für maximale Breite und Wiederverwertungsmöglichkeiten profiliert.
6. Welche Regionen deckt die gesamte Exom-Sequenzierung ab?
WES konzentriert sich auf die Exom-Erfassung von protein-kodierenden Regionen (Zielgröße typischerweise im Bereich von mehreren Megabasen, abhängig vom Erfassungsdesign), wobei die Abdeckung je nach GC-reichen oder schwer zu erfassenden Zielen variiert.
7. Welche QC-Metriken werden für WES berichtet?
Wir berichten über wichtige Qualitätskontrollen zur Abdeckung und Erfassung, wie z.B. die Leistung auf dem Ziel, Duplikation und die Verteilung der Abdeckung (einschließlich allgemeiner Zusammenfassungen der Abdeckungsgrenzen über die Ziele hinweg), um Ihnen zu helfen, die Nutzbarkeit der Daten schnell zu bewerten.
8. Welche Dateien werde ich von einem WES-Projekt erhalten?
Typische Liefergegenstände umfassen FASTQ (rohe Reads), BAM/CRAM (ausgerichtete Reads), VCF (Variantencalls), sowie eine annotierte Variantentabelle und ein prägnantes QC-/Berichtspaket (RUO).
9. Können WES-Ergebnisse für den Vergleich auf Kohortenebene verwendet werden?
Ja. Für Mehrfachstudien können Ausgaben in einem konsistenten Format und (optional) mit kohortenbewusster Analyse erzeugt werden, um die Vergleichbarkeit zwischen den Proben zu verbessern und batchbezogene Interpretationsprobleme zu reduzieren.
Referenz:
- Kyung Kim et al. Einfluss der Tiefe der Next-Generation Exom-Sequenzierung auf die Entdeckung diagnostischer Varianten. Genomik & Informatik2015, Jun; 13(2): 31–39.
Fallstudien zur gesamten Exomsequenzierung
Erste missense Mutation außerhalb des SERAC1-Lipase-Domains, die den intrazellulären Cholesterintransport beeinflusst.
Journal: Neurogenetik
Impact-Faktor: 3,269
Online veröffentlicht: 7. Oktober 2015
Zusammenfassung
MEGDEL-Syndrom ist ein seltener angeborener Stoffwechselfehler. Dieses Syndrom wurde mit Mutationen im SERAC1-Gen, das die Serin-aktive Stelle enthält, in Verbindung gebracht. Die Autoren berichteten von einer neuen homozygoten Mutation im SERAC1-Gen, die durch die Ganz-Exom-Sequenzierung bei CD Genomics identifiziert wurde. Dies ist die erste Missense-Mutation außerhalb des Serin-Lipase-Domänen des Proteins, die den intrazellulären Cholesterintransport beeinflusst.
Ergebnisse
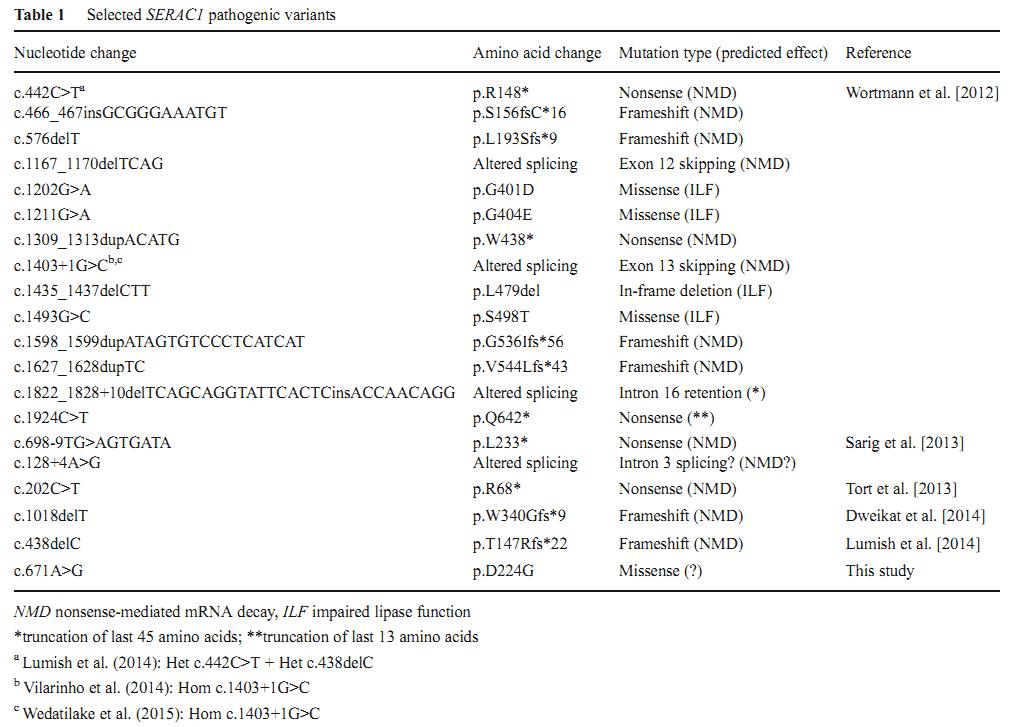
Mutationen im SERAC1-Gen
Bis heute wurden 19 Mutationen im SERAC1-Gen bei Patienten mit MEGDEL-Syndrom identifiziert (Tabelle 1). Nur drei davon sind Missense-Mutationen, die im Lipasedomäne lokalisiert sind. Die p.D224G ist die erste Missense-Mutation außerhalb der Lipasedomäne.
2. Missense-Mutation (p. D224G)
Durch die Anwendung von Whole-Exome-Sequenzierung identifizierten die Autoren eine neuartige pathogene homozygote Mutation im SERAC1-Gen. Diese Missense-Mutation veränderte eine Asparaginsäure in Glycin (Abbildung 1d). Die pathogene Rolle von p.D224G wird durch in silico-Analysen, die Erhaltung des mutierten Aminosäurerests (Abbildung 1d) und die Ansammlung von Cholesterin (Abbildung 1e) unterstützt.
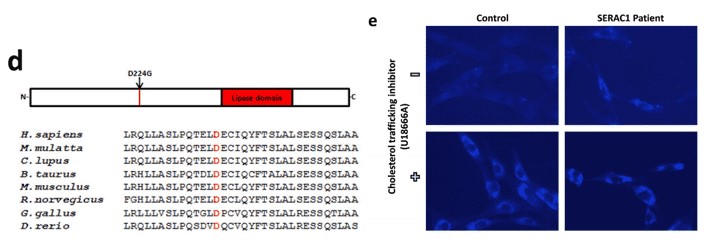 Abbildung 1. Die Position der D224-Mutation in verschiedenen Arten (d). Intrazellulärer Cholesterintransport in Fibroblasten, die von gesunden Individuen und den SERAC1-Patienten stammen. U1866A ist ein Inhibitor des Cholesterintransports.
Abbildung 1. Die Position der D224-Mutation in verschiedenen Arten (d). Intrazellulärer Cholesterintransport in Fibroblasten, die von gesunden Individuen und den SERAC1-Patienten stammen. U1866A ist ein Inhibitor des Cholesterintransports.
Referenz:
- Rodríguez-García M E, et al. Erste Missense-Mutation außerhalb des SERAC1-Lipasedomains, die den intrazellulären Cholesterintransport beeinflusst. Neurogenetik, 2016, 17(1): 51-56.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Optische Genom- und Epigenomkartierung von klarzelligem Nierenzellkarzinom
Journal: bioRxiv
Jahr: 2022
Eine unabhängige Entstehung eines jährlichen Lebenszyklus bei einer nordamerikanischen Killifischart.
Biologisches Journal der Linnean Society
Jahr: 2024
Kombinationen von Bakteriophagen sind wirksam gegen multiresistente Pseudomonas aeruginosa und erhöhen die Empfindlichkeit gegenüber Carbapenem-Antibiotika.
Journal: Viren
Jahr: 2024
Genomsequenz, Antibiotikaresistenzgene und Plasmide in einer monophasen Variante von Salmonella typhimurium, isoliert aus Einzelhandels-Schweinefleisch.
Journal: Mikrobiologie Ressourcenerklärungen
Jahr: 2024
Hochdichte-Kartierung und Kandidatengenanalyse von Pl18 und Pl20 in Sonnenblumen durch Whole-Genome-Resequenzierung
Zeitschrift: Internationale Zeitschrift für Molekulare Wissenschaften
Jahr: 2020
Identifizierung von Faktoren, die für die m6A mRNA-Methylierung in Arabidopsis erforderlich sind, zeigt eine Rolle für die konservierte E3-Ubiquitin-Ligase HAKAI.
Zeitschrift: New Phytologist
Jahr: 2017
Mehr ansehen Artikel, die von unseren Kunden veröffentlicht wurden.


