
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Bevölkerungsentwicklung
Was ist die Theorie der Bevölkerungsentwicklung?
Die Theorie der Populationsentwicklung untersucht die dynamische Gestaltung der genetischen Variation innerhalb von Populationen im Laufe der Zeit, die durch eine Reihe von evolutionären Prozessen vorangetrieben wird. Diese komplexe Theorie vereint Prinzipien aus der Genetik, Ökologie und Evolutionsbiologie, um zu erläutern, wie sich Populationen an ihre Umwelt anpassen, evolutionäre Veränderungen durchlaufen und möglicherweise in neue Arten divergieren.
Die Theorie basiert auf mehreren grundlegenden Mechanismen, die entscheidend für das Verständnis der Populationsentwicklung sind:
- Natürliche SelektionDieser grundlegende Prozess umfasst das unterschiedliche Überleben und die Fortpflanzung von Individuen basierend auf phänotypischen Variationen. Über aufeinanderfolgende Generationen hinweg werden Merkmale, die Überlebens- und Fortpflanzungsvorteile bieten, innerhalb der Population zunehmend verbreitet, wodurch ihr evolutionärer Weg gelenkt wird.
- Genetische DriftBesonders ausgeprägt in kleinen Populationen bezieht sich der genetische Drift auf zufällige Schwankungen in den Allelfrequenzen. Dieser stochastische Prozess kann zu erheblichen evolutionären Veränderungen führen, einschließlich der Fixierung oder des Verlusts von Allelen, unabhängig von ihrem selektiven Wert. Folglich kann genetischer Drift zu divergierenden evolutionären Ergebnissen führen, die unabhängig von der natürlichen Selektion sind.
- GenflussAuch bekannt als Migration umfasst der Genfluss den Transfer von Allelen zwischen Populationen. Diese Bewegung genetischen Materials kann neuartige Allele in eine Population einführen, die genetische Vielfalt erhöhen und ihren evolutionären Verlauf beeinflussen. Der Genfluss dient als Kanal für den genetischen Austausch, der entweder die Divergenz zwischen Populationen einschränken oder fördern kann.
- MutationMutationen, definiert als Veränderungen in DNA-Sequenzen, sind die Hauptquelle neuer genetischer Variationen. Diese genetischen Veränderungen können das Auftreten neuer Merkmale fördern und bieten das grundlegende Rohmaterial, das für die natürliche Selektion und andere evolutionäre Prozesse benötigt wird. Folglich sind Mutationen unerlässlich für die kontinuierliche Evolution und das Anpassungspotenzial von Populationen.
Einführung in die Analyse der Populationsentwicklung
Die Analyse der Bevölkerungsentwicklung umfasst die Anwendung modernster genomischer Technologien zur Untersuchung der genetischen Zusammensetzung und evolutionären Wege von Populationen. Dabei werden Daten genutzt, die aus Hochdurchsatz-Sequenzierung Techniken, diese Analyse zeigt genetische Variationen wie Einzel-Nukleotid-Polymorphismen (SNPs), Insertionen und Deletionen (InDels), strukturelle Variationen (SVs) und Kopienzahlvariationen (CNVs).
Schlüsselelemente der Analyse der Bevölkerungsentwicklung
- Probenentnahme und -vorbereitungDie Grundlage einer fundierten Analyse liegt in der sorgfältigen Auswahl repräsentativer Proben aus verschiedenen Teilpopulationen oder ökologischen Nischen. Die Gewährleistung der Qualität und Vielfalt dieser Proben ist entscheidend, um genaue und aussagekräftige Ergebnisse zu erzielen.
- Genomsequenzierung: Nutzung fortschrittlicher Sequenzierungstechnologien, wie z. B. Whole-Genome-Sequenzierung (WGS) und gezielte Neusequenzierung können Forscher umfangreiche genomische Daten sammeln. Zum Beispiel nutzt CD Genomics sowohl Plattformen der zweiten Generation (Illumina) als auch der dritten Generation (PacBio und Nanopore), um detaillierte genetische Informationen zu extrahieren und eine umfassende Analyse zu ermöglichen.
- VariantenkennungDie Identifizierung genetischer Variationen innerhalb von Populationen ist ein Grundpfeiler des Verständnisses evolutionärer Prozesse. Methoden zur Variantenentdeckung umfassen SNP-Calling, InDel-Erkennung sowie die Analyse von SVs und CNVs. Diese Ansätze sind entscheidend für die Kartierung genetischer Vielfalt und die Nachverfolgung evolutionärer Geschichten.
- Bioinformatische AnalyseComputationaltools und statistische Modelle spielen eine entscheidende Rolle bei der Interpretation genetischer Daten. Dies umfasst die Bestimmung der Populationsstruktur und die Identifizierung von Selektionssignaturen durch Bioinformatik-Software, die für die Variantenannotation, die Metriken der Populationsgenetik und die evolutionäre Modellierung entwickelt wurde. Solche Werkzeuge ermöglichen ein tieferes Verständnis der genetischen Grundlagen und der evolutionären Dynamik innerhalb von Populationen.
Vorteile der Analyse der Populationsentwicklung
- Umfassende genetische EinblickeEnthüllt die genetischen Grundlagen von Anpassung und Artenbildung durch die Analyse genetischer Variation in verschiedenen Populationen.
- Verbessertes Verständnis der evolutionären DynamikVerfolgt genetische Veränderungen im Laufe der Zeit, um Migrationsmuster, Anpassungsmechanismen und die Auswirkungen historischer Ereignisse aufzudecken.
- Praktische Anwendungen in der Erhaltung und ZuchtHilft dabei, genetisch vielfältige Individuen für Zucht und Erhaltung zu identifizieren und gewährleistet genetische Gesundheit und Widerstandsfähigkeit.
- Fortgeschrittene technologische IntegrationNutzt modernste Technologien und bioinformatische Werkzeuge, wobei CD Genomics präzise und hochwertige Daten für umfassende Forschungen bereitstellt.
- Umfangreiche Multiplexing-Flexibilität und Hochdurchsatz-Sequenzierung ermöglichen die Erkennung einer großen Anzahl von SNPs, InDels, CNVs und CVs.
- Zeit- und kosteneffizient.
- Widmen Sie Unterstützung von spezialisierten Wissenschaftlern auf Doktoratsniveau.
Anwendungen der Populationsentwicklung
- Forschung zum Mechanismus der künstlichen DomestikationDie genetische Beziehung zwischen Wildtyp- und domestizierten Populationen wurde durch genetische Analysen der beiden Populationen abgeleitet, anschließend wurden wichtige Gene identifiziert, die mit bedeutenden wirtschaftlichen Merkmalen in Zusammenhang stehen, was eine gute Orientierung für die landwirtschaftliche Zucht von Tieren und Pflanzen bietet.
- Analyse des Mechanismus der natürlichen SelektionDie im adaptiven Evolutionsprozess ausgewählten Gene können durch die Forschung an Populationen aus verschiedenen geografischen Gebieten entdeckt werden, wodurch umweltangepasste genetische Ressourcen für die Zuchtarbeit bereitgestellt werden.
- BevölkerungsgeschichtsforschungDurch die Analyse der möglichen Herkunft der Arten und der genetischen Variationsinformationen der Population in jedem Verbreitungsgebiet kann der Evolutionsprozess der Arten untersucht werden.
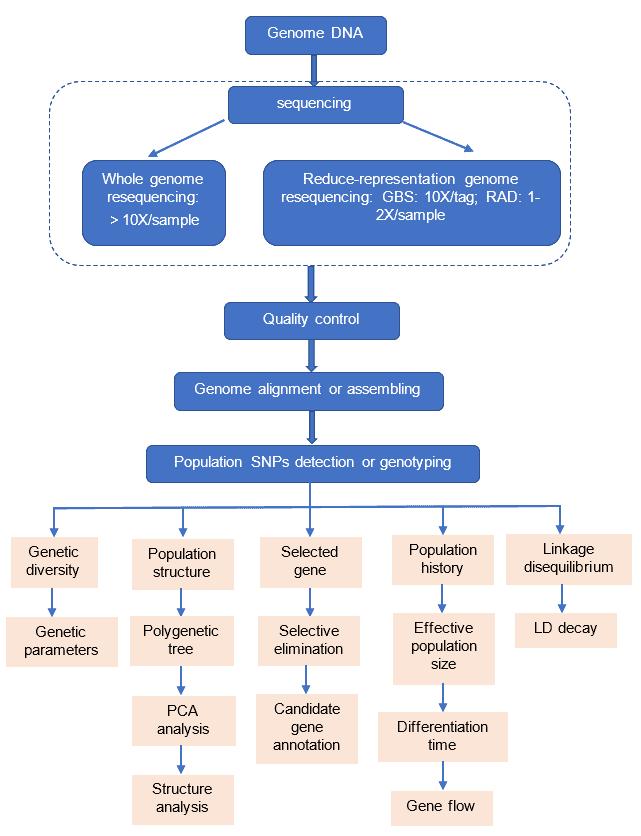
Bevölkerungsentwicklungs-Workflow
CD Genomics bietet Dienstleistungen zur Populationsentwicklung an, die die Probenentnahme umfassen, DNA-Sequenzierungund umfassende bioinformatische Analysen. Diese Dienstleistungen bewerten genetische Variation, Anpassungsmechanismen und evolutionäre Dynamiken. Durch detaillierte Dateninterpretation bieten sie wichtige Einblicke in die Populationsgeschichte, genetische Vielfalt und Erhaltungsstrategien und unterstützen die Bemühungen, die genetische Gesundheit und Biodiversität zu erhalten.

Dienstspezifikationen
Beispielanforderungen
|
|
 Klicken |
Sequenzierungsstrategie
|
 |
Bioinformatische Analyse Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an:
|
Analyse-Pipeline
Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details zur Bevölkerungsentwicklung für Ihr Schreiben (Anpassung)
Referenzen
- Nadeau N J, Ruiz M, Salazar P, et al. Populationsgenomik paralleler Hybridzonen bei den mimetischen Schmetterlingen, H. melpomene und H. erato. Genomforschung2014, 24(8): 1316-1333.
- Tine M, Kuhl H, Gagnaire P A, et al. Das Genom des Europäischen Seebarsches und seine Variationen geben Einblicke in die Anpassung an Euryhalinität und Speziation. Naturkommunikationen, 2014, 5.
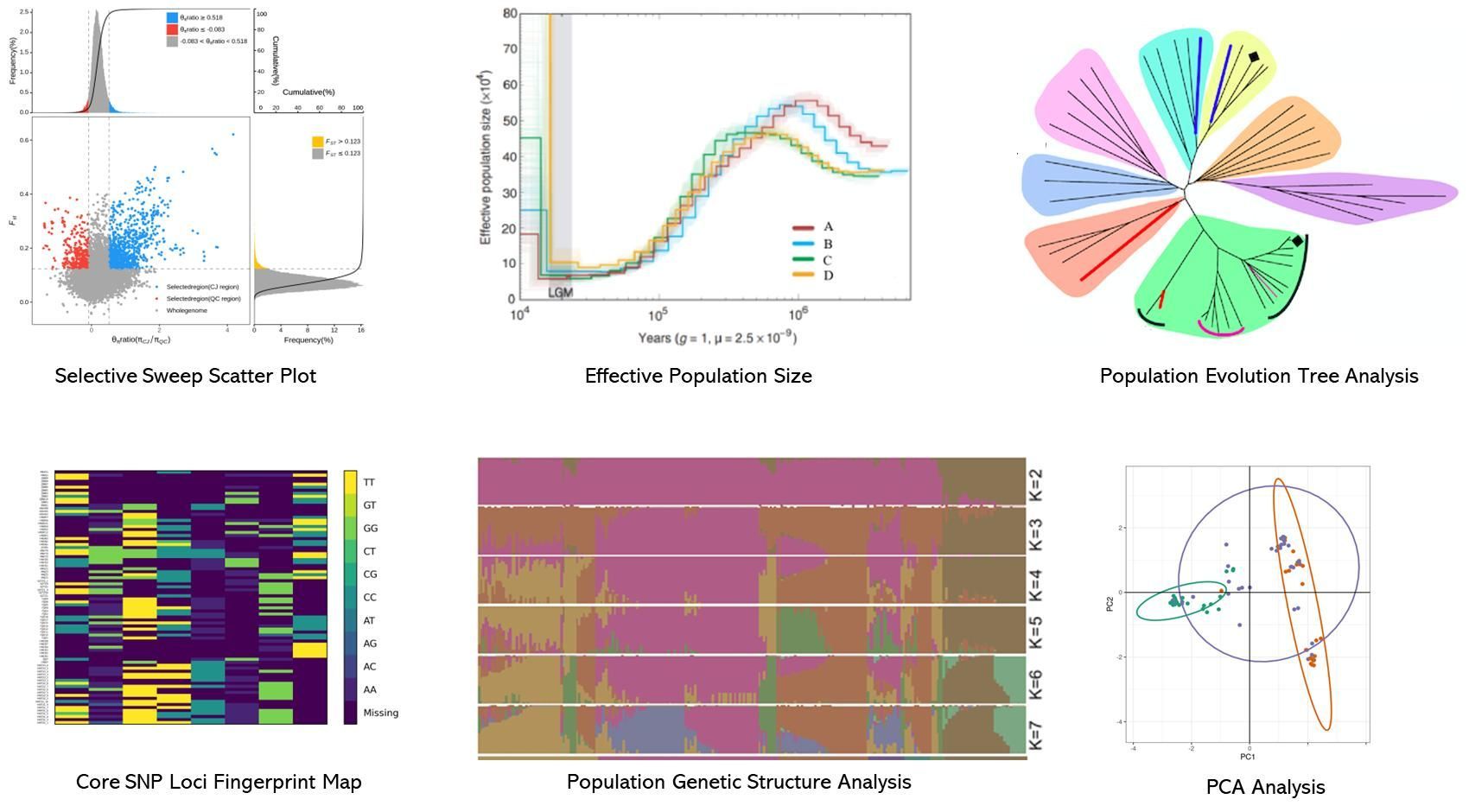
Demonstrationsergebnisse
Teilweise Ergebnisse sind unten aufgeführt:
Häufig gestellte Fragen zur Bevölkerungsentwicklung
1. Wie kann die Differenzierung von Subpopulationen abgeleitet werden?
Die Differenzierung von Subpopulationen kann durch sorgfältige Analyse geografischer und umweltbedingter Variationen abgeleitet werden. Längere geografische Isolation oder signifikante Umweltunterschiede führen häufig zur Bildung von distincten Subpopulationen. Darüber hinaus spielen Faktoren wie genetische Drift und selektive Drücke eine entscheidende Rolle bei der Förderung der Differenzierung, was zu einzigartigen genetischen Signaturen innerhalb jeder Subpopulation führt.
2. Was ist die empfohlene Sequenzierungstiefe für Studien zur Populationsentwicklung?
Um präzise und umfassende Ergebnisse in Studien zur Populationsentwicklung zu erzielen, wird eine Sequenzierungstiefe von mindestens 10X empfohlen. Höhere Tiefen, wie 30X oder 50X, können jedoch für spezifische Analysen, wie die Erkennung von strukturellen Varianten oder detaillierte evolutionäre Untersuchungen, erforderlich sein. Diese erhöhten Tiefen verbessern die Genauigkeit der Identifizierung genetischer Variationen und erhöhen die Zuverlässigkeit der Ergebnisse der Studie.
3. Welche Kriterien sollten für die Auswahl von Forschungspopulationen verwendet werden?
Die Auswahl der Forschungsbevölkerungen sollte auf klaren und unterscheidbaren Merkmalen der Subpopulationen basieren, wie den Unterschieden zwischen wilden und domestizierten Typen oder Populationen, die aus verschiedenen geografischen Regionen stammen. Es ist unerlässlich, repräsentative Proben aus jeder Subpopulation zu gewinnen, um die Genauigkeit und Validität der Analyse sicherzustellen. Die Schaffung klarer Unterscheidungen zwischen Subpopulationen verbessert unser Verständnis von genetischer Variation und evolutionären Dynamiken.
Fallstudien zur Bevölkerungsentwicklung
Die Analyse von 427 Genomen offenbart die Populationsstruktur von Moso-Bambus und die genetischen Grundlagen von Eigenschaftsmerkmalen.
Zeitschrift: Nature Communications
Impact-Faktor: 17,7
Veröffentlicht: 15. September 2021
Hintergrund
Moso-BambusPhyllostachys edulis), die 74 % der globalen Bambusflächen abdeckt und für Chinas Wirtschaft von entscheidender Bedeutung ist, sieht sich aufgrund von Umwelt- und menschlichen Einflüssen Wachstumsherausforderungen gegenüber. Whole-Genome-Resequenzierung (WGRS) Von 427 Proben aus 15 Regionen hilft dabei, genetische Variationen aufzudecken und Einblicke in die evolutionäre Geschichte sowie Merkmale, die für die Eigenschaften von Bambus relevant sind, zu gewinnen.
Materialien & Methoden
Probenvorbereitung
- Moso-Bambus
- Junge Blätter
- DNA-Extraktion
Sequenzierung
- Whole-Genome-Resequenzierung
- Illumina-Sequenzierungsplattform
- SNP- und InDel-Erkennung
- SV- und CNV-Erkennung
- Die Analyse verwandter Gene
- Phylogenie-Konstruktion
- Analyse der Bevölkerungsstruktur
- Genomweite Assoziationsstudie
Ergebnisse
Die großangelegte Whole-Genome-Resequenzierung (WGRS) von 427 Moso-Bambusproben aus 15 Regionen ergab eine geringe genomische Vielfalt, mit einer durchschnittlichen SNP-Dichte von einem SNP pro 351 Basenpaaren. Die meisten SNPs wurden in intergenen Regionen gefunden, mit einem höheren Verhältnis von nicht-synonymen zu synonymen Substitutionen im Vergleich zu anderen Pflanzen. Diese geringe Vielfalt deutet auf eine kleine effektive Populationsgröße und einen begrenzten genetischen Pool für zukünftige Züchtungen hin.
 Abb. 1: Die Landschaft der Probenahme und Varianten in sequenzierten Moso-Bambusindividuen.
Abb. 1: Die Landschaft der Probenahme und Varianten in sequenzierten Moso-Bambusindividuen.
Die balancierende Selektion spielt eine Schlüsselrolle bei der Anpassung von Moso-Bambus an seine Umwelt, indem sie die genetische Vielfalt aufrechterhält, trotz geringer insgesamt Divergenz. Die Analyse identifizierte 83 signifikante genomische Regionen, die mit balancierender Selektion in Verbindung stehen, wobei Gene, die an Krankheitsresistenz und Umweltreaktionen beteiligt sind, hochfrequente Variationen aufweisen. Dies deutet darauf hin, dass die balancierende Selektion dazu beiträgt, die genetische Vielfalt zu erhalten und die Anpassung zu unterstützen.
 Abb. 2: Die balancierende Selektion in der Moso-Bambuspopulation liegt der Anpassung zugrunde.
Abb. 2: Die balancierende Selektion in der Moso-Bambuspopulation liegt der Anpassung zugrunde.
Eine GWAS von Moso-Bambus enthüllte genetische Varianten, die mit Eigenschaften wie Halshöhe und mechanischer Festigkeit assoziiert sind. Analysierte SNPs identifizierten signifikante Marker, die mit Eigenschaften der Zellwand und der Anpassung an die Umwelt verbunden sind. Schlüssengene wie Cinnamoyl-CoA-Reduktase beeinflussen Eigenschaften wie Ligninlevels und bieten Einblicke für Zucht- und genetische Forschung.
 Abb. 3: GWAS wichtiger Eigenschaftsmerkmale.
Abb. 3: GWAS wichtiger Eigenschaftsmerkmale.
Fazit
Die Ganzgenomsequenzierung von Moso-Bambus hat eine geringe genetische Vielfalt, aber eine hohe Heterozygotie aufgrund der asexuellen Fortpflanzung offenbart. Der Naturschutz sollte sich auf vielfältige Populationen konzentrieren, und das Verständnis genetischer Variationen kann Züchtung und nachhaltige Waldwirtschaft verbessern.
Referenz
- Zhao H, Sun S, Ding Y, et al. Analyse von 427 Genomen zeigt die Populationsstruktur von Moso-Bambus und die genetischen Grundlagen der Eigenschaftsmerkmale. Naturwissenschaften Kommunikation2021, 12(1):5466.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Sammlung genetischer Daten in ethnisch basierten Studien zu Aymaras, Quechuas und Mestizen: die Herausforderungen der Genetik von Alzheimer in der peruanischen Bevölkerung (GAPP)-Studie
Zeitschrift: Alzheimer & Demenz
Jahr: 2022
Bewertung von Plasma-Biomarkern für die A/T/N-Klassifikation der Alzheimer-Krankheit bei Erwachsenen karibischer hispanischer Ethnie
Journal: JAMA Netzwerk Offen
Jahr: 2023
Erhöhte Produktion von pathogenen, luftgetragenen Pilzsporen bei der Exposition einer Bodenmykobiota gegenüber chlorierten aromatischen Kohlenwasserstoffschadstoffen
Journal: Mikrobiologie Spektrum
Jahr: 2023
Eine Splice-Variante im SLC16A8-Gen führt zu einem Defizit beim Laktattransport in aus menschlichen iPS-Zellen abgeleiteten retinalen Pigmentepithelzellen.
Journal: Zellen
Jahr: 2021
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


