
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben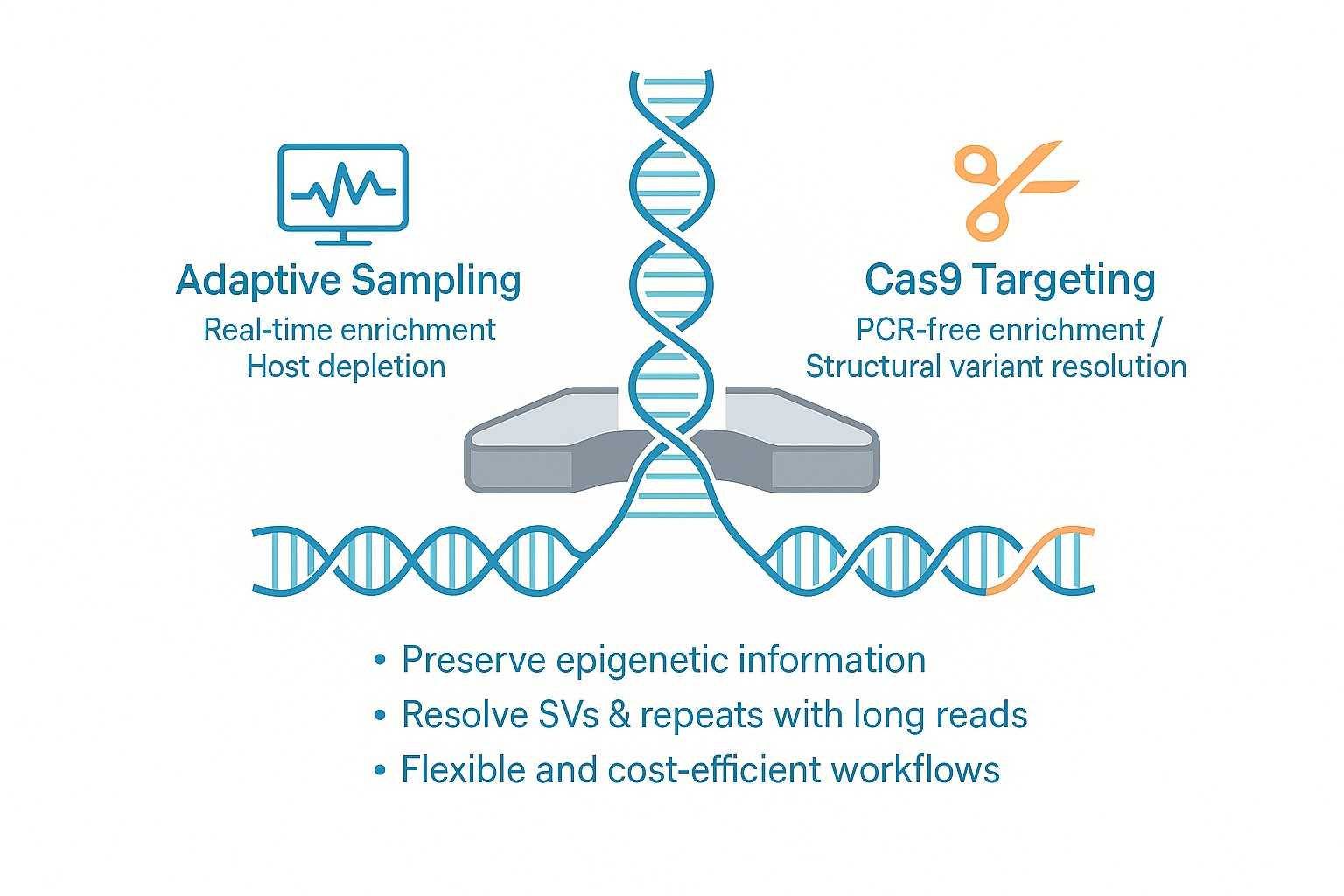
Einführung in die Nanoporen-gezielte Sequenzierung
Nanopore gezielte Sequenzierung ermöglicht es Forschern, den Sequenzierungsaufwand auf die relevantesten Teile des Genoms zu konzentrieren – sei es auf spezifische Gene, Regionen mit hoher biologischer Bedeutung oder seltene mikrobielle Taxa – und dabei unnötige Sequenzierungen von Hintergrund-DNA zu vermeiden.
Im Gegensatz zu traditionellen Methoden, die erfordern PCR-Amplifikation oder HybridisierungsfängungNanopore-Technologie bietet flexible Strategien zur Durchführung von gezielter Sequenzierung:
Nanopore-Amplikationsansätze für kosteneffektives Profiling von PCR-Produkten
Cas9-gezielte Sequenzierung für die präzise Anreicherung von krankheits- oder AMR-bezogenen Genen
Adaptive Samplingeine einzigartige softwaregestützte Methode, die DNA-Fragmente während der Sequenzierung ohne zusätzliche Labor Schritte anreichert oder verringert
Diese Flexibilität macht Nanopore-gezielte Sequenzierung besonders leistungsstark für Anwendungen in der Onkologie, Mikrobiologie, Epigenetik und Evolutionsforschung.
Warum Nanopore-Targeted-Sequenzierung wählen?
Unvoreingenommene, Langzeit-Sequenzierung
Nanopore-Sequenzierung unterstützt jede Leselänge von 50 bp bis >4 Mb, die die Möglichkeit bieten, große strukturelle Varianten, sich wiederholende Elemente und GC-reiche Regionen zu erfassen, die mit Methoden für Kurzlesungen nicht aufgelöst werden können.
Epigenetische Informationen bewahren
Im Gegensatz zur PCR- oder Sonden-basierten Anreicherung analysiert die gezielte Nanoporen-Sequenzierung direkt native DNA- und RNA-Moleküle, wobei Methylierungs- und Basenmodifikationssignale erhalten bleiben, die für die regulatorische und Krebsforschung entscheidend sind.
Echtzeit-Anreicherung
Adaptive Sampling ermöglicht es Forschern, die Datenausgabe während des Laufs zu überwachen und in Echtzeit interessante Reads zu bereichern oder zu reduzieren. Dies verkürzt die Durchlaufzeit und sorgt für eine effiziente Nutzung der Sequenzierungskapazität.
Flexible, kosteneffiziente Arbeitsabläufe
Ohne teure Erfassungsplatten oder umfangreiche Optimierung bietet die nanopore-targeted Sequenzierung einen schnelleren und skalierbareren Weg zu Ergebnissen im Vergleich zu herkömmlichen Methoden.
| Funktion | Nanoporen-gezielte Erfassung | Nanopore-Ganzgenom | Illumina gezielte Erfassung |
| Leseumfang | Lange Artikel | Lange Artikel | Kurze Texte |
| Variation | Komplexe strukturelle Variationen epigenetische Modifikationen |
Whole-Genome-Variationen epigenetische Modifikationen |
SNVs/Indels |
| Echtzeit-Sequenzierung | Ja | Ja | Nein |
| Verstärkungsbias | Nein | Nein | Ja |
| Portabilität | Hoch | Hoch | Niedrig |
| Durchsatz | Moderat | Moderat | Hoch |
| Kosten | Moderat | Hoch | Moderat |
Unsere Nanopore-Zielgerichteten Sequenzierungsdienstleistungen-Pakete
Bei CD Genomics bieten wir sorgfältig gestaltete Dienstleistungspakete für Nanopore gezielte Sequenzierung um unterschiedlichen Forschungsanforderungen gerecht zu werden. Jedes Paket fällt unter das Dach der gezielten Sequenzierung – der Auswahl spezifischer genomischer Regionen von Interesse – unterscheidet sich jedoch in den Methoden, den Eingabebedürfnissen und den gelieferten Daten. Überprüfen Sie die Optionen unten und wählen Sie das Paket aus, das am besten zu Ihren Projektzielen, der Probenqualität und Ihrem Budget passt.
| Paketname | Am besten geeignet für / Anwendungsfälle | Eingabe- und Musteranforderungen | Was ist enthalten | Vorteile |
|---|---|---|---|---|
| Basis-Amplicon-Paket | Projekte, die sich auf spezifische Hotspot-Mutationen, kleine Gen-Panels, 16S / ITS-Barcoding und die Identifizierung mikrobieller Marker konzentrieren. | DNA ≥ 50–100 ng; akzeptable Fragmentgröße ≥ ~500-1000 bp; Ziele gut bekannt; moderate Anzahl von Loci | PCR-Primerdesign; PCR-Amplifikation; Nanoporen-Sequenzierung von Amplicons; SNV / kleine Insertionen und Deletionen Aufruf; grundlegender QC-Bericht | Niedrige Kosten; schnelle Bearbeitungszeit; gut geeignet für Labore mit begrenztem Probenmaterial; hohe Tiefe über wenige Ziele |
| Cas9 / Panel Deep-Target-Paket | Mittel- bis große Genpanels; Erkennung struktureller Varianten; herausfordernde oder sich wiederholende Regionen; Erhaltung von Methylierungs- und epigenetischen Merkmalen | Hochwertige DNA (≥ 200-500 ng), bevorzugte Fragmentgröße (über 10-20 kb); minimale Degradation; ausreichende Probenreinheit | Leit-RNA-Design; Cas9-vermittelte Anreicherung; Langzeit-Nanoporen-Sequenzierung; SNV + SV-Erkennung; optionale Methylierungsprofilierung; detaillierter Bericht | Probe-freies Targeting schwieriger Regionen; behält native DNA-Modifikationen bei; in der Lage, große Insertionen / Deletionen und mobile Elemente zu erkennen |
| Adaptives Sampling-Paket | Projekte, die Flexibilität benötigen: Zielkoordinaten dynamisch ändern; Wirts-/Hintergrund-DNA-Depletion; seltene Ziele in gemischten Proben | Genomisches DNA von guter Qualität; die Menge variiert je nach Zielgröße; die Eingabebedarf hängt vom Anteil des anvisierten Genoms ab; BED-Datei mit Zielkoordinaten bereitgestellt. | Einrichtung der adaptiven Probenahme in MinKNOW; Sequenzierung mit On-Device-Anreicherung oder -Depletion; Echtzeitüberwachung; SNV-, SV- und Basismodifikationsanalyse; QC- und Abdeckungsstatistiken. | Keine zusätzliche Anreicherung im Feuchtlabor; schnelle Einrichtung; dynamisches Targeting; gut für Umwelt- oder metagenomische Proben mit Hintergrund-DNA-Problemen. |
| Umfassendes Panel + Multi-Omikationen Paket | Testen vieler Gene (z. B. erbliche Krebs-Panels), Integration von Mutations-, Struktur-, Methylierungs- und Haplotypdaten; große Studien, die ein vollständiges Funktionsset erfordern. | Hohe Eingabemenge (≥ ~500 ng–1 µg, abhängig von den Zielen); hohe DNA-Integrität; lange Fragmente sind stark wünschenswert; Proben-QC erforderlich | Vollständige Panelanreicherung (über Cas9 oder hybride Sondendesigns); Langzeitdaten; SNV / SV / Indel-Call; DNA-Methylierung / epigenetische Modifikationen; Haplotyp-Phasierung; vollständige QC / Annotation; umfassende Berichterstattung | Reiche Daten in einem Experiment; deckt alle Variantentypen und epigenetischen Marker ab; nützlich für tiefgehende Krankheitsgenetik, Merkmalskartierung, funktionelle Genomik. |
Welches Paket sollten Sie wählen?
- Wenn Ihre Ziele wenige, gut charakterisiert sind und Sie schnelle Ergebnisse benötigen, wird das Basic Amplicon Package oft ausreichen.
- Wenn strukturelle Varianten, Wiederholungen oder schwierige genomische Regionen von Bedeutung sind, wählen Sie das Cas9 / Panel Deep-Target oder das Comprehensive Panel + Multi-Omics-Paket.
- Wenn die Probe gemischt ist / Hintergrund-DNA hoch ist / erwarten Sie, die Ziele anzupassen → Adaptive Sampling bietet mehr Flexibilität.
- Für Projekte, die alle Ebenen der Einsicht (Mutationen + Struktur + Methylierung + Haplotypen) erfordern, bietet das umfassende Paket den vollständigsten Datensatz.
Anwendungen der gezielten Nanoporen-Sequenzierung
Krebsgenomik
- Erkennen Sie strukturelle Varianten wie große Insertionen, Deletionen und Inversionen mit Auflösungsgenauigkeit der Bruchpunkte.
- Profil epigenetischer Modifikationen (z. B. Hypermethylerung des MLH1-Promotors in der Lynch-Syndrom-Forschung)
- Unterstützung von erblichen Krebsrisikopanelen mit über 250 Genen, einschließlich BRCA1/2
- Aktivieren Sie die Haplotype-Phasierung für allelspezifische Risikobewertung.
Antimikrobielle Resistenz (AMR)
- Verfolgen Sie ARGs wie blaCTX-M und blaTEM mit Context-seq.
- Platzieren Sie Resistenzgene in ihrem genomischen Kontext (Plasmide, Transposons).
- Untersuchung des Übertrags von Antibiotikaresistenzgenen (ARGs) zwischen Menschen, Tieren und der Umwelt.
Mikrobiom und Metagenomik
- Entfernen von Wirts-DNA zur Anreicherung mikrobieller Genome
- Bereichern Sie seltene Taxa oder funktionale Gene (nifH, amoA, ARGs) für ökologische Studien.
- Stellen Sie nahezu vollständige mikrobielle Genome aus Arten mit geringer Häufigkeit zusammen.
Epigenetik und Genregulation
- Zielpromotoren, Enhancer oder Imprinting-Regionen für die direkte Methylierungsprofilierung
- Studie wiederholte Elemente wie LINE-1, um die Genomstabilität zu verstehen.
- Karte von allelspezifischen Methylierungsmustern in der Krebs- oder Entwicklungsbiologie
Alte DNA und Forensik
- Maximieren Sie die Nutzung von degradierten oder begrenzten Proben.
- Bereichern Sie mitochondriale Genome, Geschlechtsbestimmungsorte oder Artmarker.
Synthetische Biologie
- Überprüfen Sie künstliche Chromosomen, entwickelte Wege oder CRISPR-Bearbeitungen.
- Zielen Sie große Konstrukte an, um die Integrität und Einfügepunkte zu bestätigen.
Unser Nanopore Zielgerichteter Sequenzierungs-Workflow
Musterübermittlung & Qualitätskontrolle
- Akzeptieren Sie Blut, Gewebe, Zellen, mikrobielle Proben und alte DNA.
- DNA-Qualitätskontrolle: Überprüfung von Reinheit, Integrität und Konzentration.
Bibliotheksvorbereitung
- Amplicon-, Cas9-basierte oder Standard-Ligation-Protokolle.
Nanoporen-Sequenzierungslauf
- Skalierbarer Durchsatz auf GridION oder PromethION.
Echtzeitüberwachung & adaptive Probenahme
- Geräteinterne Auswahl für Ziele oder Host-Ausschöpfung.
Bioinformatikanalyse
- SNV/Indel-Analyse, SV-Erkennung, Methylierungsanalyse, Haplotyp-Phasierung.
Berichtszustellung
- Umfassender Datensatz, annotierte Varianten und veröffentlichungsbereite Ergebnisse.
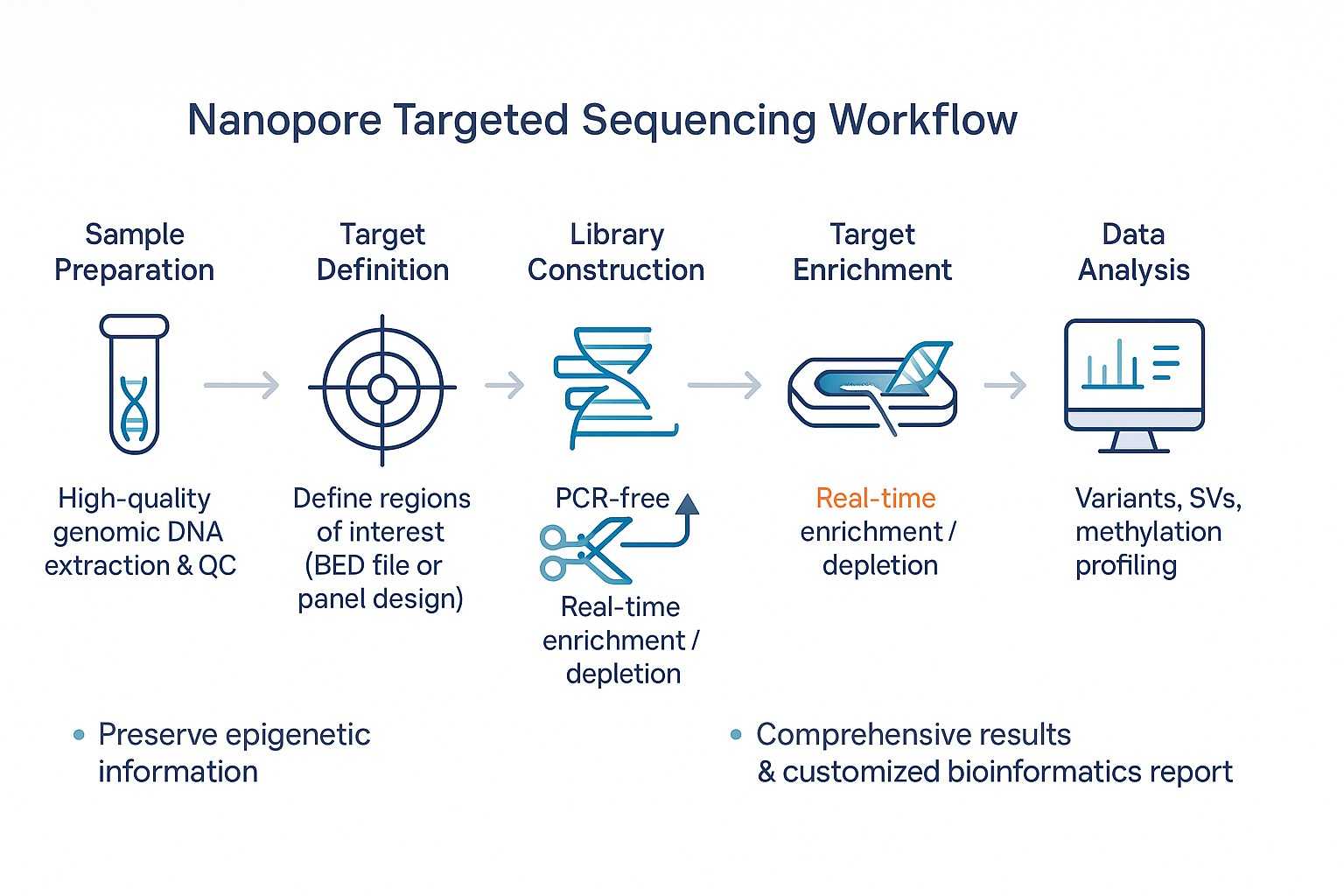 Workflow von CD Genomics Nanopore-Target-Sequenzierung, von der DNA-Extraktion und Zieldefinition über Anreicherung, Langsequenzierung bis hin zu bioinformatischen Berichten..
Workflow von CD Genomics Nanopore-Target-Sequenzierung, von der DNA-Extraktion und Zieldefinition über Anreicherung, Langsequenzierung bis hin zu bioinformatischen Berichten..
Nanopore-gezielte Sequenzierungs-Bioinformatikanalyse
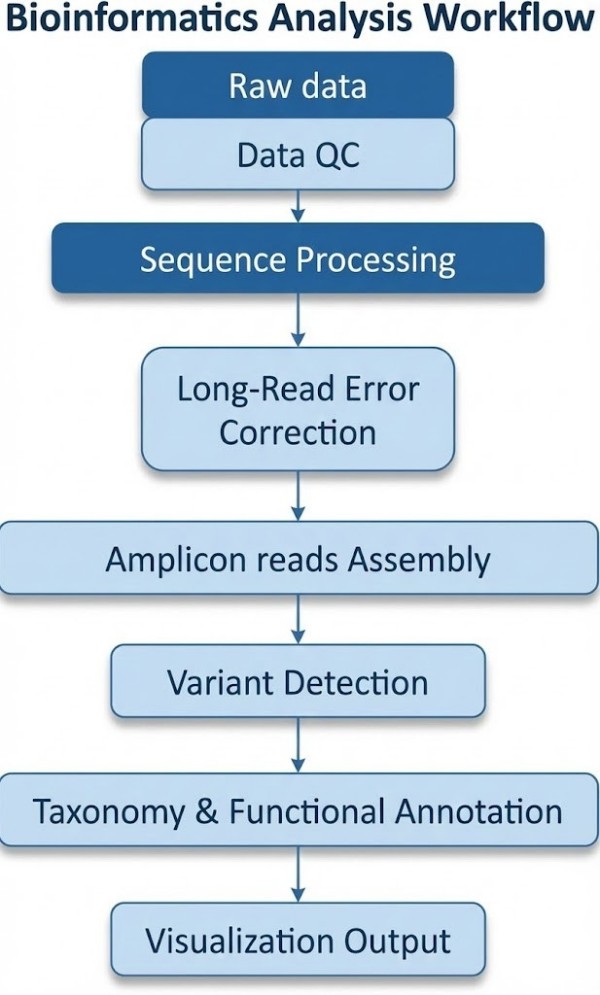
Liefergegenstände
- Rohsequenzierungsdateien (FASTQ)
- Ausrichtung (BAM) und Variantenaufrufe (VCF)
- Abdeckungs-/Anreicherungsstatistiken
- Annotierte Berichte mit SNVs, SVs, Methylierung (PDF + Excel)
- Grafische Zusammenfassungen (Circos-Diagramme, Variantenkarten, Methylierungstracks)
Warum mit CD Genomics zusammenarbeiten?
Jahre der Expertise in der Genomik-Sequenzierung über Illumina-, PacBio- und Nanopore-Plattformen
CRO-Grad Zuverlässigkeitvon Pharma-, Biotech- und akademischen Institutionen vertraut
Flexibles Projektdesignvon einzelnen Zielen zu umfassenden Krebsgen-Panels
Umfassende bioinformatische Unterstützung: Variantenannotation, Methylierungsabbildung, AMR-Genverfolgung
End-to-End-Service: Stichproben-QC, Sequenzierung, Datenanalyse, sichere Datenportal-Lieferung
Musteranforderungen
Akzeptable Probenarten & Eingaben
| Probenart | Empfohlene Menge/Eingabe | Mindestmenge/Eingabe | Mindestkonzentration / Reinheit | Hinweise zur Integrität / Handhabung |
|---|---|---|---|---|
| Hochmolekulare genomische DNA | ≥ 5 µg insgesamt, Konzentration ≥ 20-50 ng/µL | ≥ 3 µg | OD 260/280 ≈ 1,8; OD 260/230 ≈ 2,0-2,2; RNase-behandelt; frei von sichtbaren Protein-, Phenol- und Detergentienkontaminationen | Durchschnittliche Fragmentgröße groß (idealerweise ≥ 30 kb); minimale Degradation; in einem Puffer wie 10 mM Tris pH 8,0-8,4 gelagert; kühl versendet (Eispacks oder Trockeneis) |
| Mikrobielle / Umwelt-DNA | ≥ 500 ng | ≥ 300-500 ng | ≥ ~10 ng/µL; hohe Reinheit (keine Inhibitoren) | Wenn die Probe Wirte oder Kontaminanten enthält, ziehen Sie die Wirtsdepletion oder die Wahl adaptiver Probenahmewege in Betracht; während des Transports ordnungsgemäß gefroren oder stabilisiert. |
| Gewebe- oder Zellproben | Genug Material, um hochmolekulare DNA zu gewinnen (typischerweise ≥ einige µg) | So hoch wie möglich; degradierte oder niedrig-ertragende Proben können lange Reads beeinträchtigen. | Reinheit wie oben; DNA frei von RNA-/Proteinverunreinigungen; minimale DNA-Zersetzung | Frisch oder schockgefroren bevorzugt; wiederholtes Einfrieren und Auftauen vermeiden; bei Geweben helfen dünnere Scheiben; Lagerung und Transport auf Trockeneis oder blauen Eis. |
Puffer-, Lager- und Versandbedingungen
- DNA kann in RNase-freiem Wasser, TE-Puffer oder 10 mM Tris (pH 8,0-8,4) vorliegen. Vermeiden Sie Puffer mit Detergenzien, Tensiden oder hohem EDTA-Gehalt, die nachfolgende Schritte beeinträchtigen könnten.
- Für DNA in wässriger Pufferlösung oder Tris/TE, versenden Sie mit Kühlakkus oder Trockeneis, um die Kälte zu erhalten. Für DNA, die in 70% Ethanol gelagert ist, kann der Versand bei Raumtemperatur akzeptabel sein.
- Kennzeichnen Sie die Probenröhrchen deutlich: verwenden Sie einen Permanentmarker; beschriften Sie sowohl die Oberseite als auch die Seite; die Probenbezeichnungen auf den Röhrchen müssen mit denen auf dem Einreichungsformular übereinstimmen. Verwenden Sie robuste Röhrchen (1,5 mL empfohlen) oder Platten mit versiegelten Deckeln; schützen Sie vor Bruch.
- Qualitätskontrollkennzahlen
- Reinheit: OD260/280 nahe ~1,8; OD260/230 etwa 2,0-2,2. DNA-Integrität: minimale Degradation; visuelle Beurteilung auf Gel oder durch Pulsfeld oder Ähnliches; hohe Molekulargewichte (Größenverteilung verschoben zu langen Fragmenten).
- Konzentration: vorzugsweise durch fluoreszenzbasierte Methoden (z. B. Qubit) gemessen, anstatt nur durch UV-Absorption; stellen Sie sicher, dass genügend Ausgangsmaterial für das gewählte Paket vorhanden ist.
Einreichungsformular, Kennzeichnung & Dokumentation
- Vervollständigen Sie das Muster-Einreichungsformular, indem Sie die Muster-ID, den Muster-Typ, die Quantifizierung und die QC-Metriken auflisten. Die Muster-ID auf dem Formular muss genau mit dem Etikett auf dem Röhrchen übereinstimmen.
- Reichen Sie die elektronische Version der QC-Daten (z. B. Konzentration, OD-Verhältnisse, Fragmentgrößenprofil) zusammen mit physischen Proben ein.
Demonstrationsergebnisse
Verbesserung der taxonomischen Auflösung
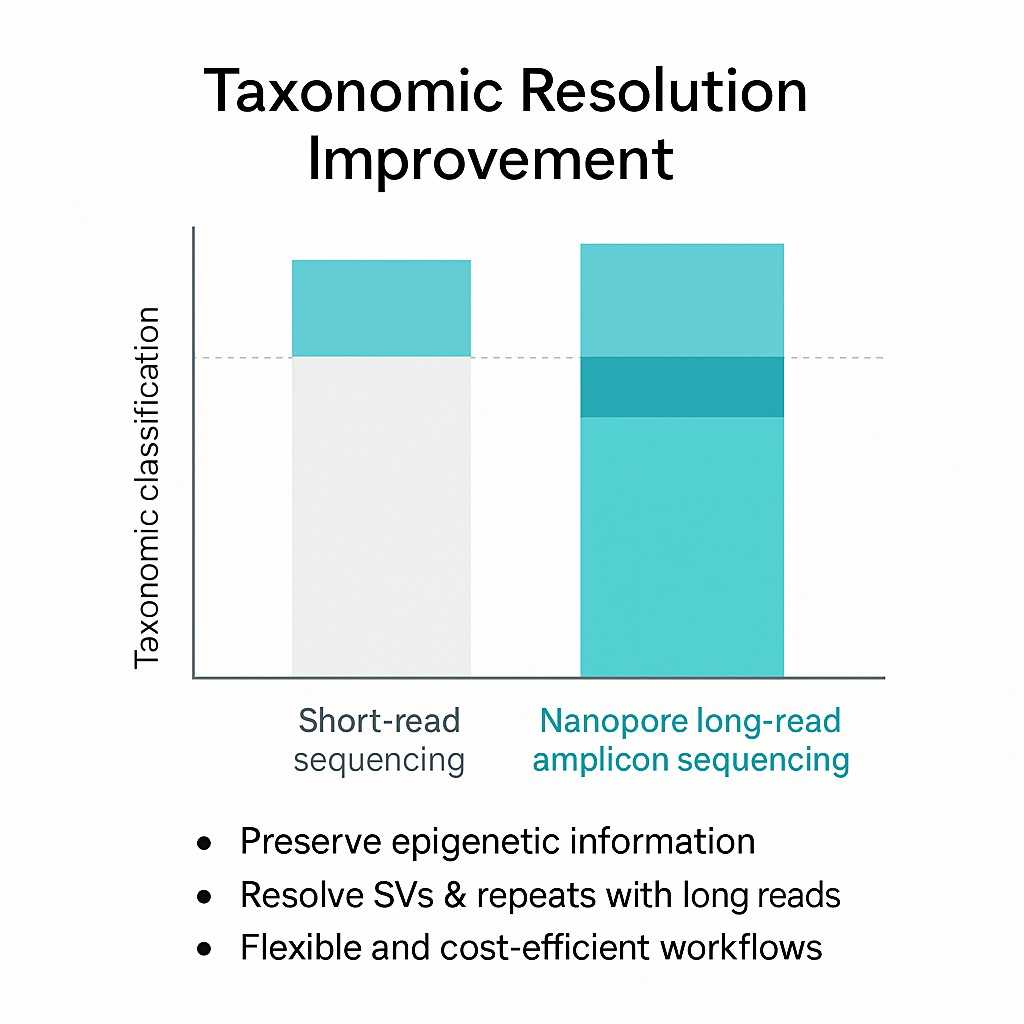
Vergleich der mikrobiellen Klassifikationsauflösung zwischen Short-Read- und Nanopore-Langread-Amplikon-Sequenzierung.
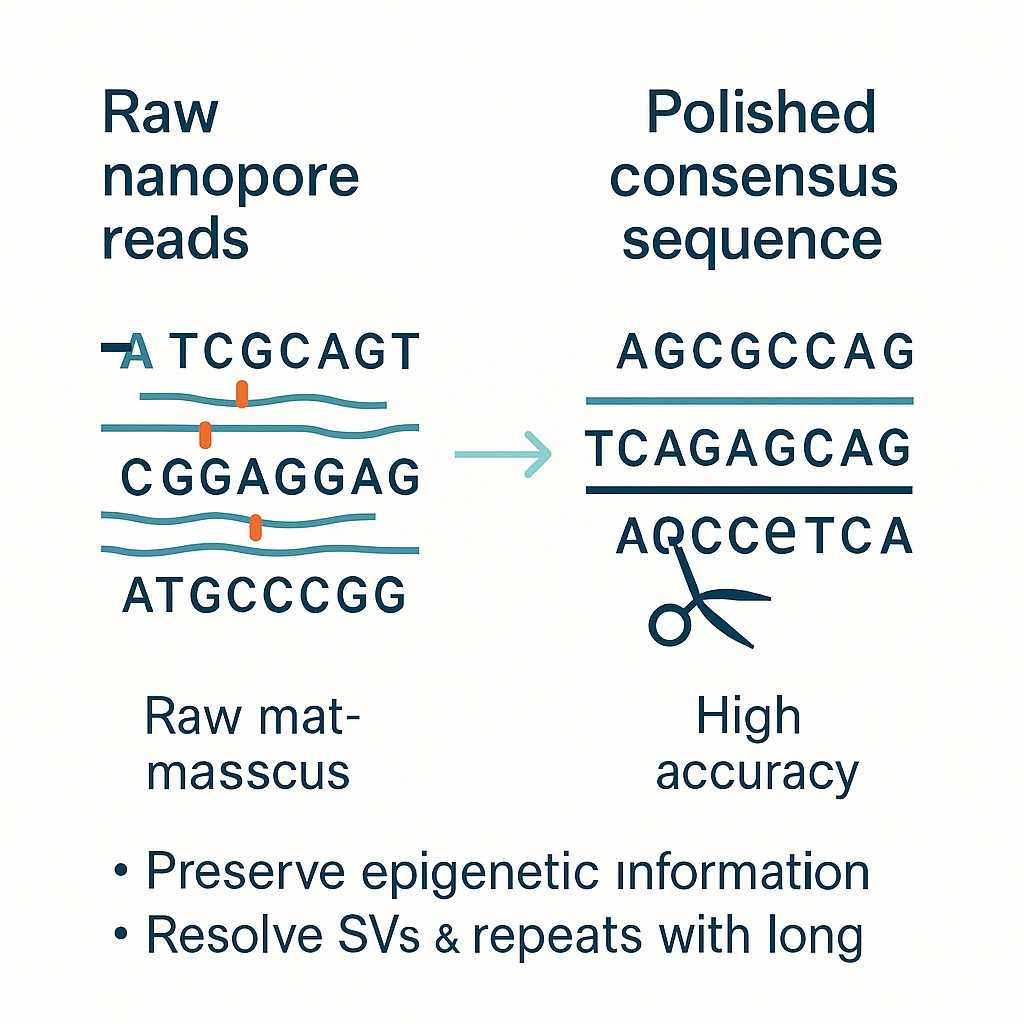
Konsensgenauigkeit mit Fehlersuppression
- Konsensuspoliierung verbessert die Genauigkeit der Variantenerkennung in nanopore-targeted Sequenzierung.
Analyse der Gemeinschaftsvielfalt
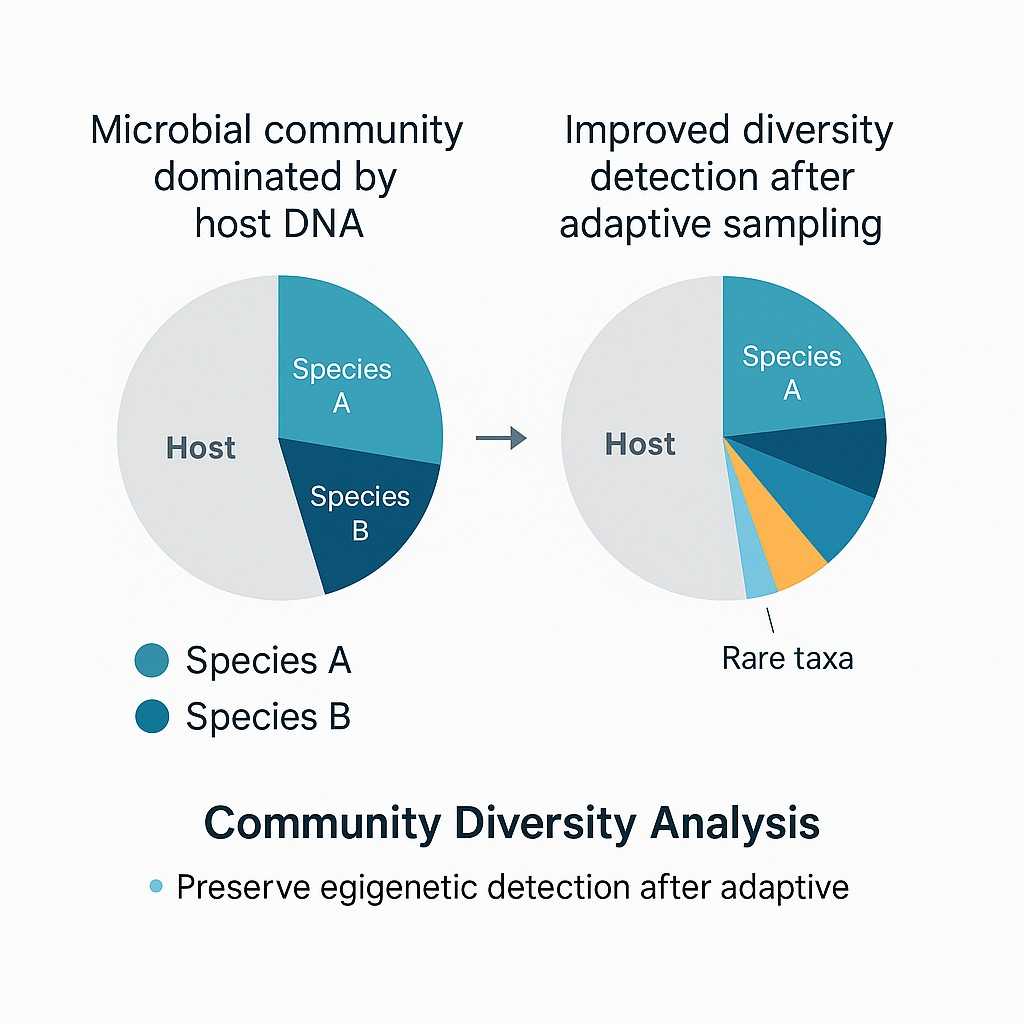
Adaptive Sampling verbessert die Erkennung seltener mikrobieller Taxa, indem die Hintergrund-DNA des Wirts reduziert wird..
Strukturelle Varianten und Wiederholungsauflösung

Cas9-zielgerichtete Anreicherung zeigt strukturelle Varianten und Wiederholungserweiterungen mit Auflösungsgenauigkeit der Bruchpunkte.
Epigenetische Erkenntnisse aus nativer DNA
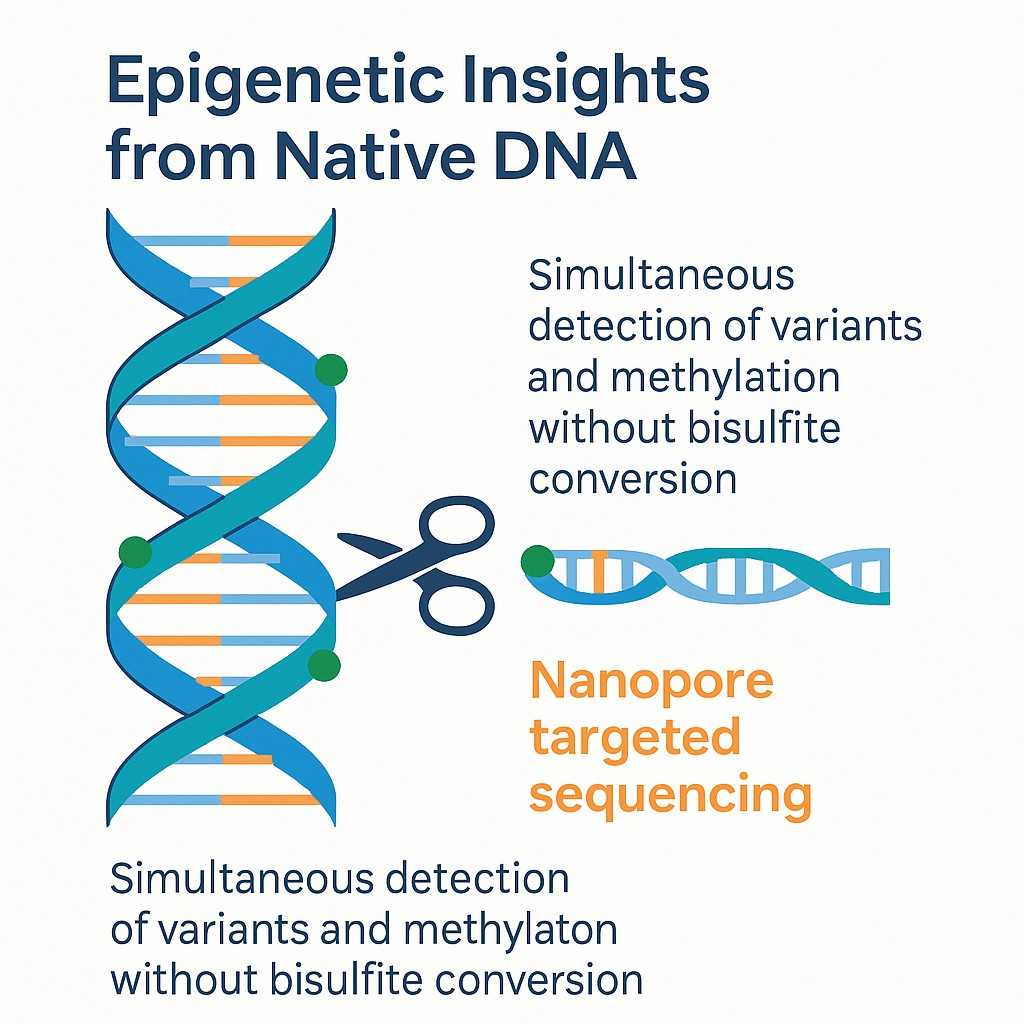
Direkte Erkennung von DNA-Methylierung während der Nanopore-gezielten Sequenzierung von nativer DNA.
Nanopore Ziel-Seq FAQs
Q1: Was ist nanopore-targeted Sequenzierung und wie unterscheidet sie sich von der Ganzgenomsequenzierung?
Die gezielte Nanopore-Sequenzierung konzentriert sich auf spezifische genomische Regionen von Interesse – wie Gene, Hotspots oder Bruchstellen struktureller Varianten – anstatt das gesamte Genom zu sequenzieren. Dieser Ansatz ermöglicht eine höhere Abdeckung dieser Ziele, weniger Datenverschwendung und die Beibehaltung langer Reads sowie epigenetischer Modifikationen. Die Ganzgenomsequenzierung bietet eine umfassende Abdeckung, ist jedoch kostspieliger und erzeugt mehr Daten, als für fokussierte Studien benötigt werden.
Q2: Welche verschiedenen Methoden oder "Routen" gibt es für das gezielte Sequenzieren mit Nanopore-Technologie?
Es gibt mehrere Ansätze: (1) PCR-basierte Amplicon-Sequenzierung für gut definierte kleine Ziele; (2) Sonden- oder Hybridfängerpaneele (weniger verbreitet bei Nanoporen, aber über maßgeschneiderte oder Drittanbieter-Designs verfügbar); (3) CRISPR-Cas9-Anreicherung, die amplifikationsfrei ist und sich für strukturelle Varianten und schwierige genomische Regionen eignet; und (4) adaptives Sampling, das in Echtzeit mithilfe von Software Reads anreichert oder depletiert, ohne zusätzliche Schritte im Labor.
Q3: Was ist der Unterschied zwischen Nanoporen-Amplicon-Sequenzierung und anderen gezielten Sequenzierungsmethoden?
Amplicon-Sequenzierung ist PCR-basiert, kostengünstig und schnell, kann jedoch Amplifikationsverzerrungen einführen, GC-reiche oder sich wiederholende Regionen übersehen und Methylierungsinformationen entfernen. Cas9-Anreicherung oder adaptive Probenahme vermeidet PCR, bewahrt Methylierung und erfasst strukturelle Komplexität über lange genomische Regionen hinweg.
Q4: Wie vergleicht sich die Nanopore-Cas9-gezielte Sequenzierung mit der Hybridfängertechnologie?
Cas9-Anreicherung ist schneller, einfacher und probe-frei und erzeugt lange Reads, die Wiederholungen und strukturelle Varianten besser auflösen. Hybrid-Capture ist nützlich für breite Panels, erfordert jedoch die Gestaltung von Proben, mehr Schritte und kann komplexe strukturelle Kontexte möglicherweise nicht so effektiv erfassen.
Q5: Kann die Nanoporen-zielgerichtete Sequenzierung epigenetische Modifikationen wie DNA-Methylierung nachweisen?
Ja. Bei der Verwendung von amplifikationsfreien Methoden wie Cas9-Anreicherung oder adaptivem Sampling erkennt die Nanoporen-Sequenzierung direkt Basismodifikationen, einschließlich Methylierung, ohne zusätzliche chemische Behandlungen oder Bibliotheksvorbereitung.
Q6: Kann adaptives Sampling auf DNA mit geringem Input oder degradierter DNA angewendet werden?
Ja, adaptives Sampling funktioniert mit standardmäßigen Nanoporenbibliotheken. Für stark fragmentierte oder niedrig-input DNA, wie FFPE oder alte Proben, können modifizierte Protokolle helfen, die Leistung zu verbessern, obwohl die Anreicherungs-effizienz von Fragmentgröße und -qualität abhängt.
Q7: Was sind die Einschränkungen oder Kompromisse der Nanoporen-zielgerichteten Sequenzierung im Vergleich zu Kurzlese- oder PCR-basierten Methoden?
Die Nanoporen-Sequenzierung kann eine niedrigere Einzel-Lese-Genauigkeit als Kurzlese-Plattformen aufweisen, obwohl die Abdeckung und der Konsens die Ergebnisse verbessern. Amplicon-Methoden können unter PCR-Bias leiden, während die Effizienz des adaptiven Samplings von der Zielgröße und der DNA-Länge abhängt. Im Gegensatz dazu liefert die Nanoporen-Sequenzierung lange Reads, bewahrt die Methylierung und löst strukturelle Varianten und Wiederholungen, die mit Kurzlese- oder nur PCR-Ansätzen nicht erfasst werden können.
Q8: Welche Probenqualität und Eingaben sind für zuverlässige Ergebnisse bei gezieltem Sequenzieren erforderlich?
DNA mit hohem Molekulargewicht wird bevorzugt, mit minimaler Degradation, angemessenen Reinheitsverhältnissen (OD 260/280 ~1,8; OD 260/230 ~2,0–2,2) und ausreichender Konzentration. Amplicon-Sequenzierung toleriert kleinere oder stärker fragmentierte Eingaben, während Cas9-Anreicherung und adaptive Probenahme am besten mit längeren DNA-Fragmenten und hochwertigen Präparaten funktionieren.
Q9: Welches Nanoporen-Gerät sollte ich für gezielte Sequenzierungsprojekte wählen?
Die Gerätee Auswahl hängt von Maßstab und Komplexität ab. MinION eignet sich für kleine Panels oder einzelne Ziele, während GridION oder PromethION für größere Panels, mehrere Proben oder Projekte mit höherem Durchsatz empfohlen werden. Multiplexing-Optionen, der Typ der Flusszelle und die Ziele für die Lese-längen beeinflussen ebenfalls die Gerätee Wahl.
Q10: Wie wird adaptives Sampling eingerichtet und was bewirkt es für gezielte Sequenzierung?
Die adaptive Probenahme wird in MinKNOW konfiguriert, indem Referenzdateien und optionale BED-Koordinaten für Zielregionen bereitgestellt werden. Während der Sequenzierung bewertet die Software die Startpunkte der Reads in Echtzeit, sodass On-Target-Reads fortgesetzt werden, während Off-Target-Reads abgelehnt werden, um Pores freizugeben. Dies ermöglicht eine Anreicherung oder Depletion ohne zusätzliche Wet-Lab-Anreicherung, wobei die Effektivität von der Zielgröße und der Qualität der Eingangs-DNA abhängt.
Q11: Wie viele Varianten können zuverlässig mit nanopore-targeted Sequenzierung nachgewiesen werden?
Nanopore-gezielte Sequenzierung kann SNVs, kleine Indels, strukturelle Varianten wie große Deletionen oder Inversionen, Wiederholungserweiterungen, wenn die Reads sie umfassen, und Haplotypen erkennen, wenn Abdeckung und Read-Länge dies zulassen. Sensitivität und Spezifität hängen vom Ziel-Design, der Sequenzierungstiefe und der DNA-Qualität ab.
Q12: Welche bioinformatischen Ergebnisse sind in einem Standardprojekt enthalten?
Typische Liefergegenstände umfassen Rohdaten (FASTQ), Ausrichtungen (BAM), Variantenaufrufe (VCF für SNVs, Indels, SVs), Abdeckungs- und Anreicherungsstatistiken, optionale Methylierungs- oder Basismodifikationsdateien sowie grafische Zusammenfassungen wie Variantenkarten oder strukturelle Variantenplots. Auch Annotationsberichte, die Ergebnisse mit Genen, bekannten Varianten oder Wegen verknüpfen, werden bereitgestellt.
Nanopore Ziel-Sequenzierungs-Fallstudien
QuelleWeiwei Gao, Chen Yang, Tianzhen Wang, Yicheng Guo, Yi Zeng (2024). Nanoporen-basierte gezielte Next-Generation-Sequenzierung von Gewebeproben zur Tuberkulose-Diagnose. Frontiers in Mikrobiologie 15:1403619. DOI: 10.3389/fmicb.2024.1403619.
1. Hintergrund
Die Diagnose von Tuberkulose (TB) im Gewebe (extrapulmonale TB, EPTB) ist schwierig, wenn kein Sputum verfügbar ist. Traditionelle Methoden (HE-Färbung + AFB, HE + PCR, HE + IHC, Xpert MTB/RIF) zeigen oft eine geringe Sensitivität bei Gewebeproben. Diese Studie untersucht, ob nanopore-basierte gezielte Next-Generation-Sequenzierung (tNGS) die diagnostische Sensitivität und Spezifität bei Gewebeproben verbessern kann, einschließlich der Erkennung potenzieller Arzneimittelresistenzen.
2. Methoden
- Insgesamt wurden 110 Gewebe-Klinikproben gesammelt.
- Der tNGS-Test zielte auf Mycobacterium tuberculosis ab. Er wurde mit HE + AFB-Färbung, HE + PCR, HE + Immunhistochemie (IHC mit anti-MPT64) und Xpert MTB/RIF verglichen.
- Ebenfalls enthalten war die Erkennung von Arzneimittelresistenzen bei Patienten, deren Proben positiv waren.
3. Ergebnisse
- tNGS erreichte eine Sensitivität von 88,2 % und eine Spezifität von 94,1 % in Gewebeproben.
- Die Vergleichsmethoden wiesen eine deutlich niedrigere Sensitivität auf: HE + AFB (30,1 %), HE + PCR (49,5 %), HE + IHC (47,3 %), Xpert MTB/RIF (59,1 %). Die Spezifitäten der Vergleichsmethoden lagen zwischen etwa 82-94 %.
- Analyse der Arzneimittelresistenz: Von 93 TB-positiven Patienten wiesen 10 (≈10,75%) eine potenzielle Arzneimittelresistenz auf, die durch tNGS identifiziert wurde.
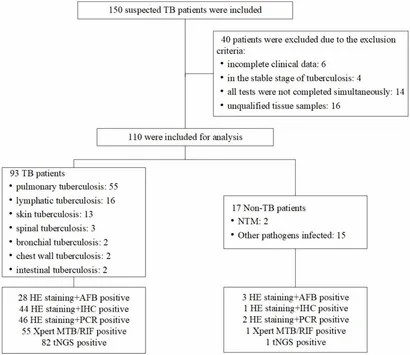 Abbildung 1. Sensitivität und Spezifität von tNGS im Vergleich zu konventionellen Diagnosetechniken für TB in Gewebeproben (HE + AFB, HE + PCR, HE + IHC, Xpert MTB/RIF).
Abbildung 1. Sensitivität und Spezifität von tNGS im Vergleich zu konventionellen Diagnosetechniken für TB in Gewebeproben (HE + AFB, HE + PCR, HE + IHC, Xpert MTB/RIF).
4. Schlussfolgerungen
Nanopore tNGS auf Gewebeproben schneidet erheblich besser ab als traditionelle Histopathologie und molekulare Methoden zur Diagnose von TB, auch in extrapulmonalen Geweben, wo die bakterielle Last gering ist. Es liefert zudem Einblicke in die Arzneimittelresistenz, was es zu einem wertvollen Werkzeug für eine schnelle und genaue Diagnose in klinischen Einrichtungen macht. Die Studie unterstützt die Verwendung von tNGS als Ergänzung zu bestehenden Diagnosetests für TB.
Referenzen:
- Gao W, Yang C, Wang T, Guo Y, Zeng Y. Nanopore-basierte gezielte Next-Generation-Sequenzierung von Gewebeproben zur Tuberkulose-DiagnoseFront Microbiol. 2024 Jul 3;15:1403619. doi: 10.3389/fmicb.2024.1403619. PMID: 39027106; PMCID: PMC11256091.
- Yang C, Gao W, Guo Y, Zeng Y. Nanopore-basierte gezielte Next-Generation-Sequenzierung (tNGS): Eine vielseitige Technologie, die auf die Erkennung von klinischen Proben mit niedriger bakterieller Last spezialisiert ist.PLoS One. 2025, 27. Mai; 20(5): e0324003. doi: 10.1371/journal.pone.0324003. PMID: 40424288; PMCID: PMC12111578.


