
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Transkriptom-Sequenzierung
CD Genomics ist stolz darauf, die meisten Arten von Transkriptomik-Dienstleistungen anzubieten und anzupassen, um die Forschungsziele und das Budget unserer Kunden zu erfüllen. Wir können Sie von der Projektplanung über den Projektstart bis hin zu hochwertigen Ergebnissen begleiten.
Was ist das Transkriptom?
Das Transkriptom ist die Gesamtheit aller RNA-Moleküle, einschließlich mRNA, rRNA, tRNA und anderer nicht-kodierender RNA, die aus einer Zelle oder einer Zellpopulation isoliert wurden. Die Transkriptomik ist das Hochdurchsatzstudium der zellulären Genexpression unter spezifischen Bedingungen, indem das vollständige Set von RNA-Transkripten, einschließlich mRNA und nicht-kodierenden RNAs, katalogisiert wird. Die Transkriptomik ermöglicht eine genomweite Analyse der Transkription mit einer Einzel-Nukleotid-Auflösung, einschließlich der Bestimmung der relativen Häufigkeit von Transkripten, der unvoreingenommenen Identifizierung alternativer Spleißereignisse und post-transkriptionaler RNA-Bearbeitungsereignisse sowie der Erkennung von Einzel-Nukleotid-Polymorphismen (SNPs). Diese Methode ist vorteilhaft gegenüber bestehenden Methoden wie Mikroarrays und Expressionssequenz-Tags. Sie ist ein leistungsfähiges Werkzeug für Studien zur frühen Embryonalentwicklung, zellulären Differenzierung, phylogenetischen Inferenz und Biomarker-Entdeckung.
Transkriptomik-Studienverfahren
Zunächst wird die gesamte RNA aus einer Probe isoliert, danach kann die Bibliotheksvorbereitung Schritte wie die Anreicherung von poly-A-basierten mRNAs oder die Anreicherung von ribosomalen-depletierten RNAs, RNA-Fragmente, die Umkehrtranskription zu cDNA, Adapterligatur und PCR-Amplifikation umfassen. Die ribosomale Depletion ist der poly-A-Selektion überlegen, da sie RNA mit oder ohne poly-A-Schwänze anreichert. Daher wird das Transkriptom, das von rRNA befreit ist, oft als gesamte RNA betrachtet, einschließlich sowohl mRNA als auch nicht-kodierender RNA. Die folgende Tiefensequenzierung erfolgt mit der Konfiguration von Illumina HiSeq (Single-End, Short-Read oder Paired-End, Long-Read) oder PacBio SMRT (Sehr lange Lese-Reaktion). Die Strategie der sehr langen Lese-Sequenzierung ist in der Lage, die vollständigen Isoformen zu sequenzieren, ohne dass eine Assemblierung erforderlich ist. Der letzte Schritt für die Transkriptomstudie ist die bioinformatische Analyse, die in der Regel die Qualitätsbearbeitung der Rohdaten, die Ausrichtung/Kartierung auf ein Referenzgenom oder von neuem Assemblierung, Nachbearbeitung, Manipulation und Qualitätskontrolle sowie fortgeschrittenes Data Mining, wie die Quantifizierung der Genexpression.
Unsere Transkriptomik-Dienstleistungen umfassen:
- RNA-SeqDie Offenlegung der RNA-Präsenz und der RNA-Expressionsniveaus unter bestimmten Bedingungen und zu bestimmten Zeitpunkten ermöglicht die Entdeckung neuer Genstrukturen, alternativ gespleißter Isoformen, Genfusionen und SNPs/InDels.
- Kleine RNA-SequenzierungProfilierung einer Vielzahl von kleinen nicht-kodierenden RNAs—miRNAs, siRNAs, piRNAs, snoRNAs, snRNAs, tRNA-Fragmente (tRFs) und mehr. Erkennung von Isoformen, Vorhersage neuer sncRNAs und Ableitung potenzieller Ziele und Funktionen.
- LncRNA-SequenzierungBieten Sie die umfassende Analyse sowohl für lncRNAs (die nicht kodierenden RNAs, die länger als 200 nt sind) als auch für mRNAs in einem einzigen Sequenzierungslauf an, um den Vergleich der Expressionsmuster von lncRNAs und mRNAs sowie die Funktionsableitung von lncRNAs und mRNAs zu ermöglichen.
- CircRNA-SequenzierungStellen Sie die Sequenzinformationen der zirkulären RNA (circRNA) mit Einzelbasenauflösung einmalig für potenzielle therapeutische und Forschungsanwendungen bereit.
- Degradom-SequenzierungAnalysiere Muster des RNA-Abbaus. Identifiziere miRNA-Spaltungsstellen aus dem Degradom effektiv und leite Zielgene genau ab.
- Bakterielle RNA-SequenzierungBieten Sie einen Prokaryoten-RNA-Sequenzierungsdienst an, um Ihre Bedürfnisse im Bereich der bakteriellen Genexpressionsprofilierung durch die Nutzung der neuesten Techniken voranzutreiben.
- Ribosomen-ProfilingStellen Sie eine Ribosomen-Profiling-Strategie bereit, die auf der Tiefensequenzierung ribosomen-geschützter mRNA-Fragmente basiert und eine genomweite Untersuchung der Translation mit sub-Kodon-Auflösung ermöglicht.
- Gesamt-RNA-SeqAnalysiere sowohl kodierende als auch mehrere Formen von nicht-kodierender RNA für einen umfassenden Überblick über das Transkriptom.
- Gezielte RNA-SequenzierungBereitstellung qualitativer und quantitativer Informationen für die Analyse der differentiellen Expression, die Messung der allelspezifischen Expression und die Verifizierung von Genfusionen.
- Exosomale RNA-SequenzierungBieten Sie den Exosomenforschern einen umfassenden Service zur Untersuchung von exosomenassoziierten RNA-Informationen an.
- ultra-niedriges Input-RNA-SequencingErmöglicht das Studium von Proben mit einer begrenzten Anzahl von Zellen oder mit einer extrem geringen Menge an Eingangs-RNA.
- Dual-RNA-SequenzierungBieten Sie direkte Einblicke in das Zusammenspiel zwischen Wirt und Erreger.
- microRNA-Sequenzierung: Speziell werden miRNAs profiliert – kleine (~21–23 nt) nicht-kodierende RNAs, die die Genexpression post-transkriptionell regulieren. Unser umfassender Service umfasst die Qualitätskontrolle der Proben, die Bibliotheksvorbereitung, Hochdurchsatz-Sequenzierung und Datenanalyse. Sie erhalten eine Quantifizierung mit Einzelbasenauflösung bekannter miRNAs, die Entdeckung neuer miRNAs und Einblicke in miRNA–mRNA-Regulationsnetzwerke. Ideal für die Entdeckung von Biomarkern und Studien zur Genregulation.
- mRNA-SequenzierungsdienstQuantifiziert Kodierungs-Transkriptome mit Illumina NovaSeq. Wir bieten Expressionsprofilierung, Erkennung von Spleißvarianten, Identifizierung von Genfusionen und Analyse von RNA-Bearbeitungen an. Zu den Dienstleistungen gehören Qualitätskontrolle, Bibliothekskonstruktion, Hochdurchsatz-Sequenzierung und fortgeschrittene Bioinformatik, die auf Ihre Projektziele zugeschnitten sind. Ideal für die Forschung zur Genexpression in Krankheitsmodellen und Pharmakogenomik.
Servicevorteile
Erkennung aller RNA-TypenDie traditionelle Transkriptom-Sequenzierung nutzt die Oligo d(T)-Methode, um RNA-Moleküle mit Poly(A)-Schwänzen zu reinigen. Viele gängige RNA-Moleküle, wie z. B. zirkuläre RNA und bestimmte lange nicht-kodierende RNAs (LncRNAs), haben jedoch keine Poly(A)-Schwänze, was zu ihrem Verlust während der Verarbeitung führt. CD Genomics bietet rRNA-Depletion-Kits an, die für verschiedene Spezies entwickelt wurden und die Erhaltung aller RNA-Typen ermöglichen.
Geeignet für degradierte ProbenCD Genomics verwendet proprietäre Kits, die mit degradierten Proben kompatibel sind und die Erhaltung von RNA-Informationen in stark degradierten Proben ermöglichen. Dieser Ansatz eignet sich besonders gut für die Transkriptomanalyse klinischer Proben.
Strangenspezifische Detektion: Viele genetische Loci produzieren Antisense-RNAs mit entscheidenden regulatorischen Funktionen, die in die entgegengesetzte Richtung transkribiert werden. Konventionelle Methoden zur Bibliotheksvorbereitung können diese Antisense-RNAs nicht unterscheiden, aber CD Genomics nutzt strand-spezifische Bibliotheksvorbereitung, die eine genaue Erkennung von Antisense-RNAs ermöglicht und somit umfassendere Informationen über das Transkriptom bereitstellt.
Spezialisierte bioinformatische Analyse: Wir verfügen über ein starkes Bioinformatik-Team, das in der Lage ist, verschiedene tiefgehende Datenanalyseanforderungen für Kunden zu erfüllen.
Hohe AuflösungEs kann ähnliche Gene innerhalb von Genfamilien und einzel-nukleotidliche Unterschiede, die durch variables Spleißen verursacht werden, erkennen.
Breite ErkennungsreichweiteEs kann präzise Kopien von wenigen bis zu zehntausenden zählen und gleichzeitig sowohl häufige als auch seltene Transkripte identifizieren und quantifizieren.
Datenanalyse
Differenzielle mRNA-, zirkuläre RNA- und lange nicht-kodierende RNA (LncRNA) Ausdrucksanalyse
Nach dem Erwerb von Hochdurchsatz-Sequenzierungsdaten wenden wir strenge Kriterien an, die einen minimalen Fold Change von ≥ 2 und ein Signifikanzniveau mit einem P-Wert von ≤ 0,05 erfordern, um differentielle exprimierte mRNA, zirkuläre RNA und LncRNA zu identifizieren.
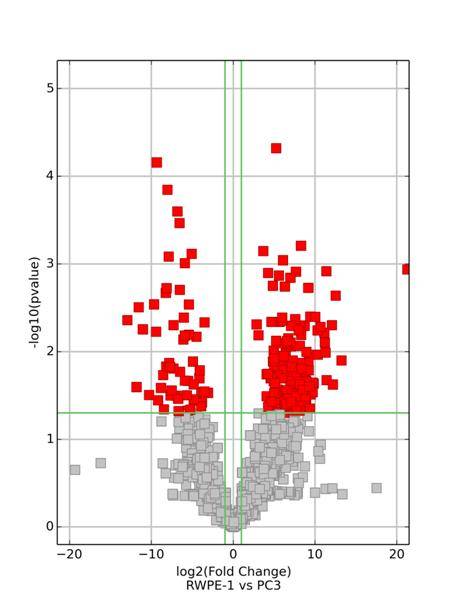
Clusterbildung von differentiell exprimierten mRNA, zirkulärer RNA und LncRNA
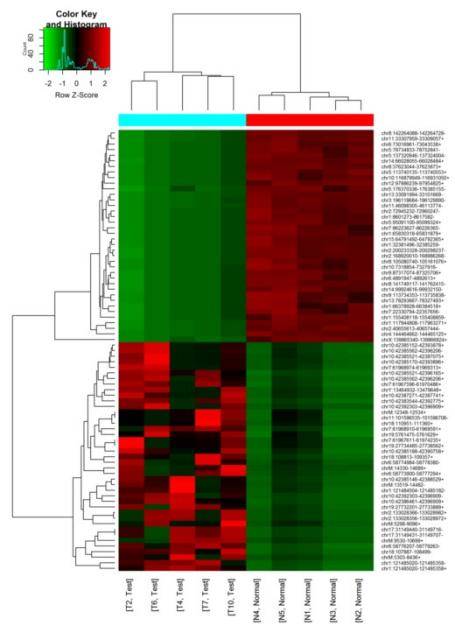
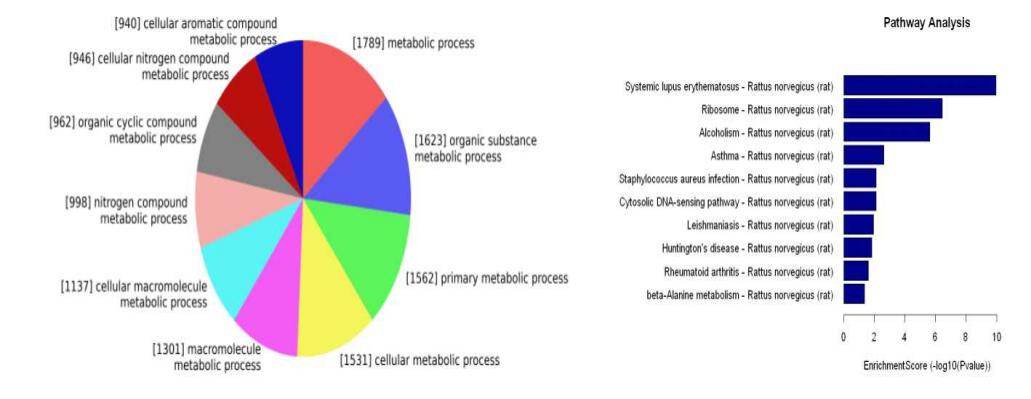
Genomontologie (GO) und Signalweg-Analyse von unterschiedlich exprimierten mRNA, zirkulärer RNA und LncRNA
Die Durchführung von funktionalen und Signalweg-Anreicherungsanalysen der Quellgene von differentially exprimierten mRNA, zirkulärer RNA und LncRNA erleichtert die Ableitung der Funktionen dieser RNA-Moleküle.
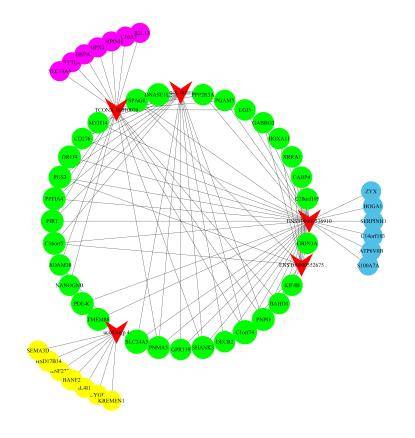
Aufbau eines Co-Expressionsnetzwerks für lange nicht-kodierende RNA und mRNA
Durch die Nutzung der Co-Expressionsassoziation zwischen langen nicht-kodierenden RNAs (LncRNAs) und messenger RNAs (mRNAs) erstellen wir ein Co-Expressionsnetzwerkdiagramm. Dieser Ansatz unterstützt die Identifizierung von Zielgenen der LncRNAs und die Ableitung der Funktionen von LncRNAs.
Wettbewerbliche endogene RNA (ceRNA) Analyse
Durch die Analyse des zirkulären RNA-microRNA-Interaktionsnetzwerks können wir dazu beitragen, die Funktion und Mechanismen von zirkulären RNAs als miRNA-Schwämme zu entschlüsseln.
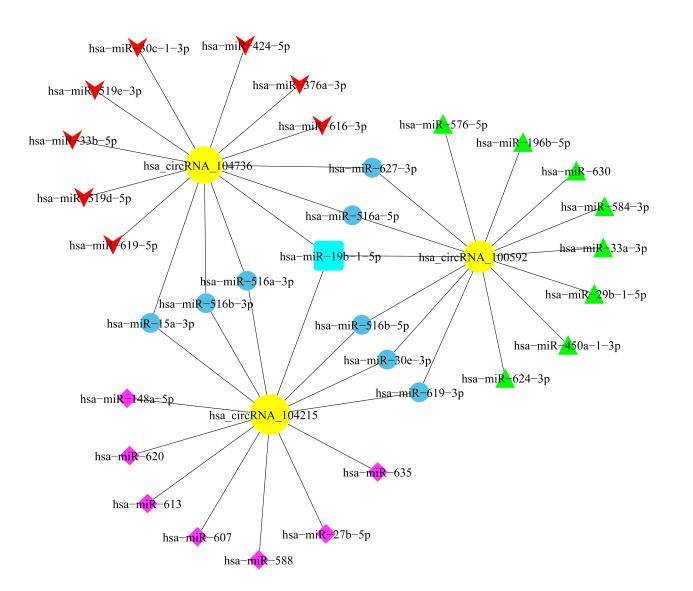
Aufbau des circRNA/LncRNA-miRNA-mRNA Netzwerks
Bestimmte circRNA/LncRNA-Moleküle können die Expression von miRNA-Zielgenen regulieren, indem sie an miRNAs binden. Durch die Analyse von circRNA/LncRNA-miRNA-mRNA-Assoziationen können wir dabei helfen, die circRNA/LncRNA-Moleküle, die als miRNA-Schwämme fungieren, und ihre Wirkungsmechanismen abzuleiten.
Unsere einzigartige Kombination aus langen und kurzen Reads, Einzel- und Paarend-Sequenzierung, Strangspezifität und der Kapazität für Zehntausende bis Milliarden von Reads pro Lauf ermöglicht es uns, Ihre Proben auf vielfältige Weise zu sequenzieren. Unser erfahrenes Personal kann Ihnen helfen, zu definieren, wie unsere Dienstleistungen am besten für Ihr Projekt genutzt werden können, und unsere strengen Qualitätskontrollen können die Integrität der gelieferten Ergebnisse gewährleisten. Bitte zögern Sie nicht, unsere Spezialisten zu kontaktieren und geeignete Strategien für die Bibliotheksvorbereitung und Sequenzierung zu besprechen.
Circular DNA tumor viruses make circular RNAs
Proceedings of the National Academy of Sciences | 2018Repeated immunization with ATRA-containing liposomal adjuvant transdifferentiates Th17 cells to a Tr1-like phenotype
Journal of Autoimmunity | 2024Role of the histone variant H2A.Z.1 in memory, transcription, and alternative splicing is mediated by lysine modification
Neuropsychopharmacology | 2024FAK loss reduces BRAFV600E-induced ERK phosphorylation to promote intestinal stemness and cecal tumor formation
Elife | 2023

