
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Bulk-Segregant-Analyse (BSA)
Was ist BSA?
BSA, Bulk-Segregant-Analyse, ist eine QTL-Kartierungstechnik zur Identifizierung genomischer Loci, die ein Merkmal von Interesse beeinflussen. Die Technik beinhaltet die Bildung von zwei Gruppen, indem Individuen mit extrem gegensätzlichen Phänotypen gekreuzt werden oder ein interessanter Mutant mit dem Wildtyp gekreuzt wird. Anschließend werden zwei Pools erstellt, indem Individuen aus den Extremen der phänotypischen Verteilung ausgewählt werden, gefolgt von Pool-Sequenzierung. Dann werden die Allelfrequenzen für die beiden Pools geschätzt; Unterschiede würden zwischen den beiden Pools auftreten, wenn die Regionen des Genoms kausale Loci enthalten.

Was ist BSA-seq?
Da die Techniken zur Gruppierung von Bevölkerungen fortgeschritten sind und die Kosten für Next-Generation Sequencing (NGS) hat erheblich abgenommen, die Integration von Whole-Genome-Resequenzierung Die Bulked Segregant Analyse (BSA) ist zu einem prominenten Ansatz geworden. Die Fusion von BSA mit NGS, die als BSA-seq bezeichnet wird, hat die Identifizierung eng verbundener Marker für wichtige Merkmale beschleunigt, wodurch die Entdeckung von Genen verbessert und die Auflösung der Kartierung von Quantitativen Trait Loci (QTL) erhöht wurde. Bemerkenswerterweise bietet BSA-seq erhebliche Zeitersparnisse beim Aufbau experimenteller Populationen, insbesondere bei der Lokalisierung von QTLs, was es zu einem schnellen und effizienten Mittel zur Identifizierung funktioneller Gene und Loci macht, die mit quantitativen Merkmalen assoziiert sind. Zusammenfassend lässt sich sagen, dass BSA-seq, das auf der Kombination von Populationsgruppierung und Technologien der zweiten Generation basiert, in der Lage ist, Stellen von Einzel-Nukleotid-Polymorphismen (SNP) zu erkennen, die mit phänotypischen Merkmalen assoziiert sind. Es hat breite Anwendung bei der präzisen Lokalisierung von QTLs und der Identifizierung funktioneller Zielgene gefunden.
Prinzipien der BSA-seq-Technologie
Die BSA-seq-Technik hat viele der Einschränkungen, die mit der Erstellung von Near Isogenic Lines (NILs) in Pflanzen verbunden sind, adressiert. Ihr Prinzip besteht darin, ein Paar von Elternlinien zu hybridisieren, die kontrastierende Merkmale von Interesse aufweisen. Anschließend werden aus einer segregierenden Nachkommenschaftspopulation (typischerweise der F2-Generation) 20 bis 50 individuelle Pflanzen ausgewählt, die extreme Phänotypen für das Zielmerkmal zeigen. Die DNA dieser Individuen wird dann extrahiert und äquimolar gepoolt, was zur Schaffung von zwei unterschiedlichen Genpools führt. Diese beiden Genpools sollten sich hinsichtlich des interessierenden Merkmals unterscheiden, während alle anderen genomischen Loci randomisiert bleiben. Polymorphe Marker, die aus dem Vergleich dieser beiden Pools identifiziert werden, können mit einem funktionalen Gen oder einem QTL von Interesse assoziiert sein.
Vorteile und Merkmale von BSA-seq
- Kurze Versuchsphase
- Genauigkeit der Kartierungsergebnisse
- Kostenwirksam
Flussdiagramm des BSA-Experiments
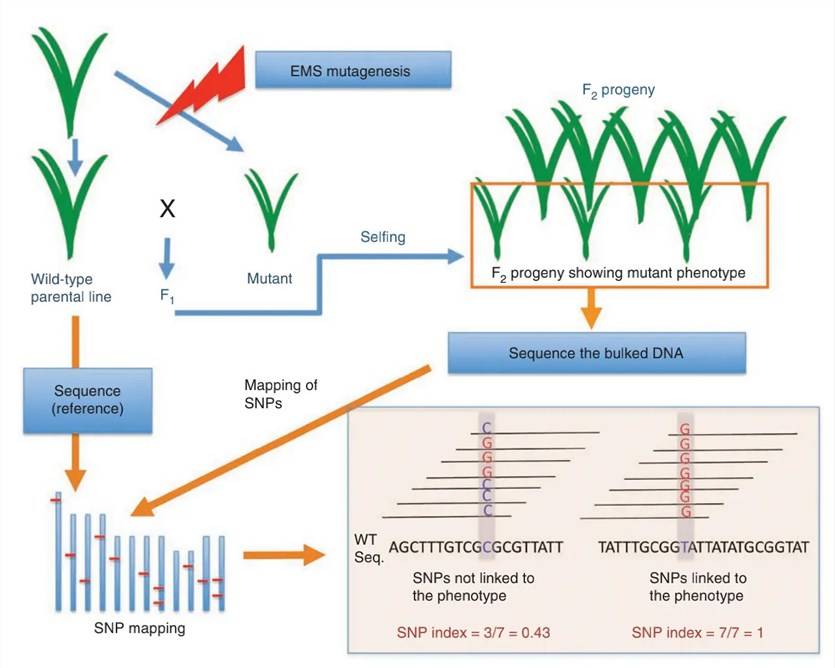 Nature Biotechnology, 2011., Akira Abe et al.
Nature Biotechnology, 2011., Akira Abe et al.
Dienstspezifikationen
Musteranforderungen
|
|
 Klicken |
Sequenzierungsstrategie
|
 |
Bioinformatikanalyse Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an:
|
BSA-seq-Datenanalyse
Um funktionale Genloci zu identifizieren, ist ein Vergleich auf SNP-Ebene zwischen den beiden gepoolten und sequenzierten Populationen unerlässlich. Die am häufigsten verwendete Methode zu diesem Zweck ist der SNP-Index-Ansatz. Sein zugrunde liegendes Prinzip umfasst die statistische Analyse der Basen an jeder Nukleotidposition unter Verwendung der Sequenzierungsreads. Ein Referenzelternteil oder ein bestehendes Referenzgenom wird typischerweise als Referenz ausgewählt. Die Anzahl der Reads im Nachkommenpool, die an einer bestimmten Nukleotidposition mit der Referenz übereinstimmen oder von ihr abweichen, wird gezählt, und das Verhältnis der abweichenden Reads zu den Gesamtreads an dieser Position wird berechnet, was den SNP-Index ergibt. Der SNP-Index wird innerhalb eines gleitenden Fensters bestimmt, normalerweise mit einer Fenstergröße von 1 Mb und einem 10 kb-Inkrement für jeden Slide. Zusätzlich wird Δ(SNP-Index) verwendet, um den Unterschied in den SNP-Indizes zwischen den beiden Genpools zu messen, wodurch Variationen an einzelnen Loci erfasst werden. Die Ergebnisse werden typischerweise in einem Manhattan-Plot visualisiert, wie unten dargestellt, wobei die horizontale Achse chromosomale Positionen darstellt, die vertikale Achse des SNP-Index-Plots die berechneten Read-Verhältnisse für jede SNP-Position angibt und die vertikale Achse des Δ(SNP-Index)-Plots den Unterschied in den SNP-Indizes zwischen den beiden gepoolten Proben darstellt. Je größer der absolute Wert von Δ(SNP-Index) ist, desto signifikanter ist ein Locus mit dem extremen Merkmal assoziiert.
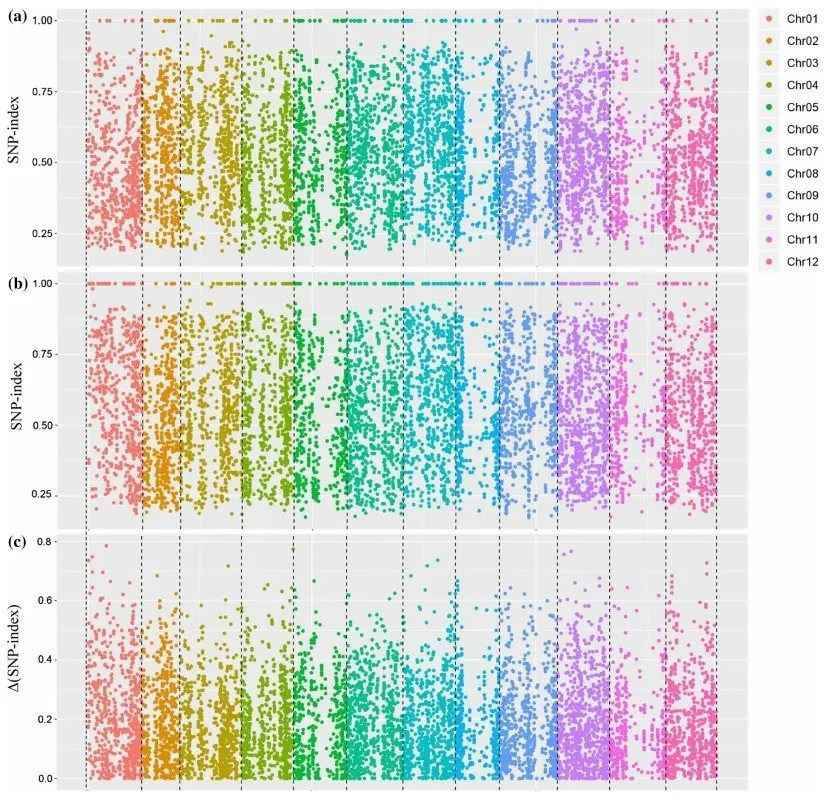 Beispiel für SNP-Index und Δ(SNP-Index) Berechnungsergebnisse
Beispiel für SNP-Index und Δ(SNP-Index) Berechnungsergebnisse
Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
Referenz
- Michelmore RW, Paran I, Kesseli RV. Identifizierung von Markern, die mit krankheitsresistenten Genen verknüpft sind, durch gebündelte Segregationsanalyse: eine schnelle Methode zur Erkennung von Markern in spezifischen genomischen Regionen unter Verwendung von segregierenden Populationen. Proc Natl Acad Sci USA1991;88(21):9828-9832. doi:10.1073/pnas.88.21.9828
- Zhang K, Li Y, Zhu W, et al. Feine Kartierung und Transkriptomanalyse des viridierenden Blattexpressors v-2 in Gurke (Cucumis sativus L.). Front Pflanzenwissenschaften. 2020;11:570817. Veröffentlicht am 25. September 2020. doi:10.3389/fpls.2020.570817
- Lee SB, Kim JE, Kim HT, Lee GM, Kim BS, Lee JM. Die genetische Kartierung des c1-Locus durch GBS-basiertes BSA-seq offenbarte Pseudo-Response Regulator 2 als Kandidatengen, das die Farbe von Paprikafrüchten steuert. Theor. Appl. Genet.. 2020;133(6):1897-1910. doi:10.1007/s00122-020-03565-5
- Guo Z, Cai L, Chen Z, et al. Identifizierung von Kandidatengenen, die die Kältetoleranz von Reis in der kalten Region in der Schossphase steuern, durch BSA-Seq und RNA-Seq. R Soc Open Sci. 2020;7(11):201081. Veröffentlicht am 18. November 2020. doi:10.1098/rsos.201081
- Zhao Z, Sheng X, Yu H, Wang J, Shen Y, Gu H. Identifizierung von Kandidatengenen, die an der Bildung von Curd-Riceyness bei Blumenkohl beteiligt sind. Int J Mol Sci. 2020;21(6):1999. Veröffentlicht am 15. März 2020. doi:10.3390/ijms21061999
- Aguado E, García A, Iglesias-Moya J, et al. Kartierung eines partiellen Andromonoecy-Locus in Citrullus lanatus unter Verwendung von BSA-Seq- und GWAS-Ansätzen. Front Pflanzenwissenschaften. 2020;11:1243. Veröffentlicht am 19. August 2020. doi:10.3389/fpls.2020.01243
Demonstrationsergebnisse
Teilweise Ergebnisse sind unten aufgeführt:

Häufig gestellte Fragen zur Bulk-Segregant-Analyse
Q1: Welche Art von Elterntypen eignet sich für den Aufbau von Populationen?
A1: Es wird empfohlen, Elterntypen mit minimalen heterozygoten Loci und einzelnen Merkmalsunterschieden auszuwählen. Darüber hinaus sollten die Unterschiede zwischen den Elterntypen nicht zu groß sein, da übermäßige Differenzierung zu ausgeprägten falsch positiven Ergebnissen führen kann, was es schwierig macht, die tatsächlichen Zielregionen zu identifizieren. Bei der Auswahl von Elterntypen für das Zielmerkmal können verschiedene Methoden eingesetzt werden, wie z.B. EMS-Mutagenese, natürliche Individuen und UV-Mutagenese, die alle in der Bulk Segregant Analyse (BSA) verwendet werden können.
Q2: Was rechtfertigt die Bevorzugung der Auswahl einzelner Linien gegenüber gemischten Populationen, natürlichen Populationen oder Baum-Populationen?
A2: Gemischte Populationen, natürliche Populationen und Baum-Populationen sind oft durch einen signifikanten Grad an Heterozygosität gekennzeichnet, was zu einer erheblichen genetischen Vielfalt führt. Selbst innerhalb begrenzter DNA-Regionen ist das Zusammenleben mehrerer allelischer Genotypen ein häufiges Vorkommen. Diese Häufigkeit unterschiedlicher Genotypen innerhalb von DNA-Pools führt zu einer verringerten Zuverlässigkeit bei der Erkennung von Einzel-Nukleotid-Polymorphismen (SNPs) und der genauen Berechnung von Genotypfrequenzen.
In hybriden Linien enthält ein DNA-Segment in der Regel nur zwei Allele, eines von jedem Elternteil, was die Ausrichtung der Reads an einer Referenzsequenz und die Variantenerkennung relativ unkompliziert macht. In hoch heterozygoten Populationen erhöht das Pooling von DNA aus verschiedenen Quellen die Vielfalt der Reads, was zu höheren Fehlerquoten bei der Ausrichtung an Referenzgenomen und der SNP-Erkennung führt.
In der BSA-seq-Analyse basierend auf den hybriden Linien sind die meisten mutierten Genotypen hochfrequent (≥0,5), was ihre Erkennung relativ einfach macht. Im Gegensatz dazu weisen natürliche Populationen oft zahlreiche niederfrequente SNPs auf, und wenn sie zusammengefasst werden, wird es schwierig, echte niederfrequente SNPs von solchen zu unterscheiden, die durch Sequenzierungs- oder Alignierungsfehler entstehen, was die Datenanalyse kompliziert.
Q3: Ist es machbar, elterliche DNA aus Samplingsproben und Nachkommendna aus Blattproben zu extrahieren?
A3: Die Auswahl spezifischer Pflanzenbestandteile für die DNA-Extraktion hängt von zwei entscheidenden Faktoren ab: der Zusammensetzung der Samenschale und des Endosperms sowie dem Grad der Homozygotie. Der Ursprung, der überwiegend dem mütterlichen Elternteil innerhalb der Samenschale zugeschrieben wird, kann einen Einfluss ausüben. Ebenso ist der Grad der Homozygotie bei den Nachkommen eine wichtige Überlegung. In Fällen, in denen die Homozygotie erheblich ist und der Unterschied zwischen der DNA der Eltern und der Nachkommen minimal ist, wird der Einfluss geringer.
Q4: Was ist das empfohlene Verfahren zur Poolung von Nachkommen-DNA?
A4: Es ist ratsam, DNA aus jeder Nachkommensprobe einzeln zu extrahieren und diese anschließend in äquimolaren Mengen zu kombinieren. Dieser Ansatz dient dazu, Hintergrundgeräusche zu minimieren und potenzielle systematische Fehler zu verringern.
Q5: Welche Kriterien sollten Nachkommenpopulationen erfüllen?
A5: Grundsätzlich kann jede hybride Nachkommenschaft, die eine Merkmalssegregation für das Zielmerkmal zeigt, für die Bulk Segregant Analyse (BSA) geeignet sein. Häufig verwendete Populationen sind F2-Generationen, Rückkreuzpopulationen und rekombinante Inzuchtlinien. Bei qualitativen Merkmalen kann die Nachkommenschaft ein Verhältnis von 1:1 oder 3:1 aufweisen, abhängig vom spezifischen untersuchten Merkmal. Im Kontext quantitativer Merkmale ist es vorteilhaft, wenn die Nachkommenschaft Merkmale zeigt, die einer Normalverteilung entsprechen. Bei ausgeprägten Abweichungen von der Normalität ist es wichtig, den potenziellen Einfluss von rezessiven letalen Genen zu bewerten.
Q6: Ist es möglich, die Größe des Kandidatenbereichs und die Anzahl der Kandidatengene sicherzustellen?
A6: Die Dimensionen der Kandidatenregion und die Anzahl der Kandidatengene hängen von verschiedenen Faktoren ab, einschließlich der Populationsgröße, dem Grad der genetischen Divergenz zwischen den Elterngenmaterialien, den Eigenschaften des Zielmerkmals, der Sequenzierungstiefe und dem genomischen Umfeld, das spezifisch für die untersuchte Art ist. Diese Parameter können durch die Perspektive projektspezifischer Erfahrungen und relevanter Literatur geschätzt werden.
Q7: In Fällen, in denen das abgebildete Intervall eine übermäßige Größe aufweist, welche Maßnahmen können zur Anpassung ergriffen werden?
A7: Um das Mapping-Intervall zu verfeinern, kann man Anpassungen am Konfidenzintervall vornehmen oder die Auswahl spezifischer SNP- und InDel-Marker innerhalb des BSA-Mapping-Intervalls vornehmen, um anschließend eine lokale Karte zu erstellen. Diese Strategie erweist sich als effektiv, um den Umfang des Mapping-Bereichs zu minimieren.
Q8: Wie wird das Zielgen nach der Auswahl der Kandidatengene validiert?
A8: Zu den gängigen Validierungsmethoden gehören:
SNP-Validierung:
Konvertieren Sie Kandidaten-SNPs in CAPS- oder dCAPS-Marker zur Validierung.
Analysiere die Erkennungsstellen von Restriktionsenzymen für potenzielle SNPs, wähle SNPs aus, die Änderungen in den Erkennungsstellen der Enzyme verursachen, amplifiziere die entsprechenden Fragmente mit SNPs unter Verwendung spezifischer Primer und führe dann eine Enzymverdauung sowie eine Gelelektrophorese durch, um SNPs in CAPS-Marker umzuwandeln. Analysiere die Polymorphie der CAPS-Marker zur Validierung.
PCR amplifizieren Sie die Kandidaten-SNP-Regionen und validieren Sie die Amplifikationsprodukte mit Sanger-Sequenzierung.
Validieren Sie die Genexpression von Kandidatengenen in verschiedenen Phänotypen mittels RT-PCR.
Führen Sie eine differenzielle Genexpressionsanalyse basierend auf Transkriptomen durch, um signifikante Unterschiede in der Genexpression zu untersuchen.
RNAi-Analyse: Verwenden Sie RNAi-Technologie, um gezielt die Expression spezifischer Gene zu silencing oder auszuschalten.
Q9: Welche Anforderungen gibt es an die Elterntypen während der Populationskonstruktion?
A9: Elterliche Linien sollten so rein wie möglich sein, und diese Reinheit kann durch Selbstbestäubung erreicht werden. Die beiden elterlichen Linien sollten signifikante Unterschiede im Zielmerkmal aufweisen, während andere Merkmale so konsistent wie möglich sein sollten, um Störungen in den nachfolgenden Mapping-Analysen zu minimieren.
Q10: Was ist die Begründung für die Empfehlung von Homozygotie in den Elternlinien für das Zielmerkmal in BSA-seq?
A10: Das grundlegende Konzept der BSA-seq Merkmalskartierung basiert auf der Identifizierung von SNPs aus beiden Elternquellen und der anschließenden Berechnung des SNP-Index über das gesamte Genom innerhalb von Nachkommenpools. Wenn die Elternlinien nicht homozygot sind, kann dies zu einer verringerten Erkennungsrate von Nachkommen-SNPs und einem verminderten SNP-Index führen. Folglich wäre es notwendig, um eine erfolgreiche Kartierung zu erreichen, die Schwelle für die Auswahl des SNP-Index zu senken, was möglicherweise zu einer erhöhten Häufigkeit von falsch-positiven Ergebnissen führen könnte. Bei der Berechnung des SNP-Index besteht die gängige Praxis darin, die Elternlinie als Referenz zu verwenden und homozygote Loci zu filtern, wobei der SNP-Index anschließend in dem Nachkommenpool an diesen spezifischen Loci berechnet wird.
Q11: Was ist die optimale Anzahl an Nachkommensproben, die gesammelt werden sollen?
A11: Die Sammlung von Nachkommensproben sollte den folgenden Richtlinien entsprechen: Für qualitative Merkmale ist es ratsam, so viele rezessive Individuen wie möglich zu sammeln, mindestens jedoch 20 Individuen, die typischerweise zwischen 30 und 50 liegen. Ebenso sollte eine gleiche Anzahl von dominanten Individuen gesammelt werden. Bezüglich quantitativer Merkmale wird allgemein empfohlen, die obersten 5%-10% der Individuen auszuwählen, die die extremsten Merkmalswerte aufweisen, um sie weiter zu bewerten. Darüber hinaus kann die Erstellung eines Histogramms basierend auf den Merkmalsdaten der Nachkommen wertvoll sein, um den Auswahlprozess zu leiten.

Q12: Welche erforderliche Sequenzierungstiefe ist für elterliche und Nachkommenlinien während der Neusequenzierung notwendig?
A12: Um die Genauigkeit von SNP- und InDel-Markern sicherzustellen, sollte das Sequencing eine bestimmte Tiefe aufweisen. Es wird empfohlen, dass die Elternlinien eine Sequierungstiefe von mindestens 20X haben. Die Sequierungstiefe für die gepoolten Proben sollte basierend auf der Anzahl der Proben bestimmt werden, mit einer durchschnittlichen Tiefe von mindestens 1X pro Probe. Zum Beispiel, wenn es 30+30 Nachkommen gibt, sollte die Sequierungstiefe für jedes Nachkommen-Pool nicht weniger als 30X betragen. Wenn es das Budget zulässt, sollten Sie in Erwägung ziehen, die Tiefe weiter zu erhöhen.
Q13: Kann die reduzierte Genomsequenzierung für die BSA-Phänotypkartierung verwendet werden?
A13: Die Technologie der reduzierten Genom-BSA kann nur 1 % bis 10 % des gesamten Genoms erfassen. Wenn die untersuchte Art ein großes Genom hat und das interessierende Merkmal von mehreren kleinen Genen mit zahlreichen assoziierten Loci kontrolliert wird, kann die reduzierte Genomsequenzierung einige Loci erfassen, wird jedoch die Mehrheit davon verpassen, was für weitere Forschungen nachteilig sein kann. Bei qualitativen Merkmalen oder quantitativen Merkmalen, die von Hauptgenen kontrolliert werden, besteht ein hohes Risiko im Zusammenhang mit reduzierter Genom-BSA. Es wird empfohlen, eine reduzierte Genomsequenzierung sowohl für die Elternlinien als auch für die Population durchzuführen und eine vollständige Genom-Verknüpfungskarte für die Kartierung der Merkmalgene zu erstellen.
Bulk-Segregant-Analyse Fallstudien
Klassischer Fall: Next-Generation Sequencing aus der Bulked-Segregant-Analyse beschleunigt die gleichzeitige Identifizierung von zwei qualitativen Genen in Sojabohnen.
Tagebuch: Grenzen der Pflanzenwissenschaften
Impact Factor: 6,627
Veröffentlicht: 31. Mai 2017
Hintergrund
Bulked-Segregant-Analyse (BSA) bieten einen unkomplizierten Ansatz zur schnellen Identifizierung von molekularen Markern, die eng mit Phänotypen assoziiert sind. BSA-Techniken wurden in verschiedenen Arten eingesetzt, um wichtige Gene zu lokalisieren. Mit den Fortschritten in der DNA-Sequenzierungstechnologie basieren BSA-Methoden auf Next-Generation Sequencing (NGS) haben die Identifizierung von krankheitsverursachenden Genen erheblich beschleunigt. Sojabohne, eines der weltweit am häufigsten angebauten Hülsenfrüchte, hat im Vergleich zu anderen Pflanzen bei der Identifizierung und Isolation von Genen zurückgelegen, was auf eine begrenzte genetische Variation, ein komplexes und großes Genom sowie eine niedrige Effizienz der genetischen Transformation zurückzuführen ist. Nur wenige Gene, die spezifische Merkmale wie das Wachstum des Stammes und die Anzahl der Samen pro Schote steuern, wurden identifiziert. Die Entwicklung von Methoden zur schnellen Identifizierung von Genen, die wichtige agronomische Merkmale kontrollieren, ist von größter Bedeutung für den Fortschritt der Forschung zur Genfunktionalität von Sojabohnen.
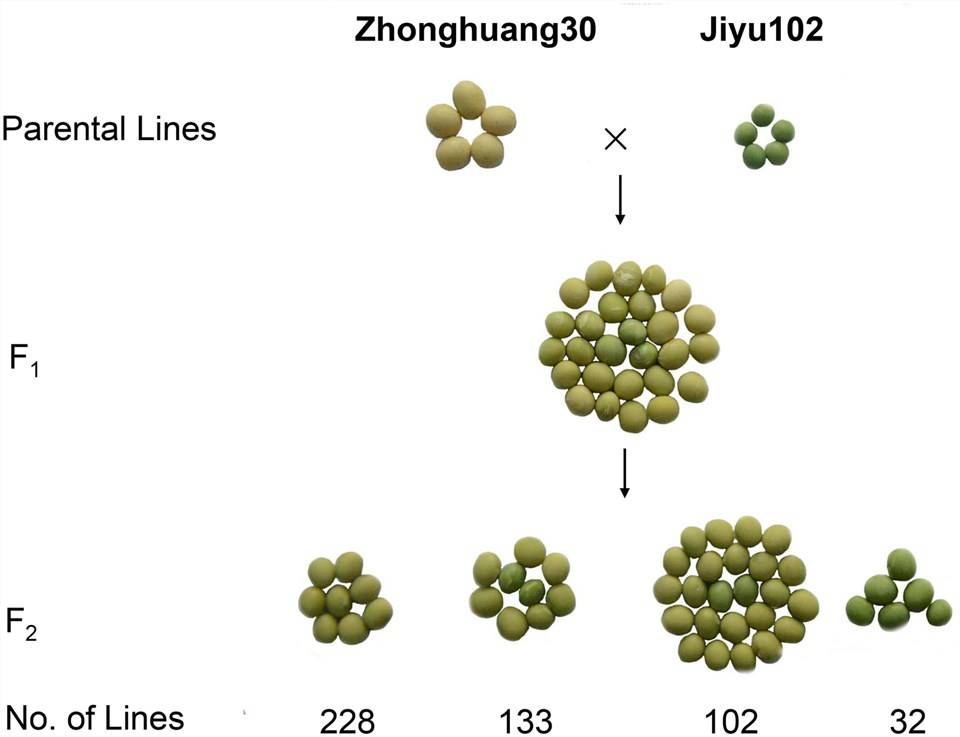 ABBILDUNG 1. Genetische Analyse der Keimblattfarbe in einem Kreuzungsexperiment zwischen ZH30 und JY102.
ABBILDUNG 1. Genetische Analyse der Keimblattfarbe in einem Kreuzungsexperiment zwischen ZH30 und JY102.
Beispielquelle: F2-Pedigree mit Fokus auf den Phänotyp der Blattfarbe gelb/grün
Stichprobengröße: 2 Elternlinien + 30 Nachkommen mit gelben Blättern + 30 Nachkommen mit grünen Blättern
Sequenzierungstiefe: Elterliche Linien (12X + 9X), Nachkommen (59X + 53X)
Materialien & Methoden
Probenvorbereitung
- Sojabohne
- Junge Blätter
- DNA-Extraktion
Sequenzierung
- Nächste Generation Sequenzierung
- Illumina HiSeqTM 2500 Plattform
- SNP-Index-Analyse
- Gleitfensteranalyse
Ergebnisse
Zhonghuang30 ist die Elternlinie mit gelben Blättern, und Jiyu102 ist die Elternlinie mit grünen Blättern. Durch Rückkreuzung der F1-Generation mit Zhonghuang30 (der Elternlinie mit gelben Blättern) trat keine Segregation auf, was die Dominanz der gelben Farbe in der Elternlinie bestätigte. Basierend auf den Segregationsverhältnissen in der F2-Generation und den F2:3-Pedigrees wurde festgestellt, dass die Blattfarbe von zwei Genen kontrolliert wird, wobei Grün ein rezessives Merkmal ist.
Mit der QTL-seq-Methode wurde die vorläufige Lokalisierung des Zielmerkmals (Blattfarbe) durchgeführt, und basierend auf dem ΔSNP-Index wurde das Zielmerkmal zunächst auf zwei Intervalle kartiert, qCC1 (~2,68 Mb) auf Chr1 und qCC2 (~2,68 Mb) auf Chr11. Anschließend wurde eine Feinkartierung unter Verwendung von SSR- und Indel-Rekombinationsmarkern an 200 Nachkommen durchgeführt, um die Kartierungsregionen einzugrenzen. qCC1 wurde auf eine Region von 30,7 kb fein kartiert, die vier Gene umfasste, wobei das D1-Gen ein Homolog des grünen Gens in Arabidopsis ist, wie in anderen Studien berichtet. qCC2 wurde auf eine Region von 67,7 kb fein kartiert, die neun Gene umfasste, und ähnlich ist eines dieser Gene, D2, ein berichtetes Homolog des grünen Gens in Arabidopsis.
Die funktionale Erkundung und Validierung wurden für diese beiden Kandidatengene durchgeführt. Es wurde festgestellt, dass das D1-Gen eine fehlende T-Basis zwischen den Elterlinien aufwies, was zu einer Frameshift-Mutation und einer vorzeitigen Beendigung in der grünen Elterlinie JY102 führte. Darüber hinaus wies das D2-Gen eine 322 bp lange Sequenzwiederholung zwischen den Elterlinien auf, was zu einer vorzeitigen Beendigung dieses Gens in der grünen Elterlinie JY102 führte.
Referenz
- Song J, Li Z, Liu Z, et al. Next-Generation-Sequencing aus einer gebündelten Segregantenanalyse beschleunigt die gleichzeitige Identifizierung von zwei qualitativen Genen in Sojabohnen. Grenzen der Pflanzenwissenschaften2017, 31;8:919.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Sammlung genetischer Daten in ethnisch basierten Studien bei Aymaras, Quechuas und Mestizen: die Herausforderungen der Genetik von Alzheimer in der peruanischen Bevölkerung (GAPP) Studie
Zeitschrift: Alzheimer & Demenz
Jahr: 2022
Bewertung von Plasma-Biomarkern für die A/T/N-Klassifikation der Alzheimer-Krankheit bei Erwachsenen karibischer hispanischer Ethnie
Journal: JAMA Netzwerk Open
Jahr: 2023
Erhöhte Produktion von pathogenen, luftgetragenen Pilzsporen bei der Exposition einer Bodenmykobiota gegenüber chlorierten aromatischen Kohlenwasserstoffschadstoffen
Journal: Mikrobiologie Spektrum
Jahr: 2023
Eine Splice-Variante im SLC16A8-Gen führt zu einem Defizit beim Laktattransport in aus menschlichen iPS-Zellen abgeleiteten retinalen Pigmentepithelzellen.
Zeitschrift: Zellen
Jahr: 2021
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


