
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Vollständige Transkript-Sequenzierung (Iso-Seq)
CD Genomics verfügt über umfangreiche Erfahrung in der Bereitstellung von Iso-Seq-Diensten, indem vollständige Transkripte ohne Zusammenstellung produziert werden. Eine strenge Qualitätskontrolle wird nach jedem Verfahren durchgeführt, um umfassende und genaue Ergebnisse zu gewährleisten.
Was ist Iso-Seq?
Isoform-Sequenzierung (Iso-Seq) ist eine Hochdurchsatz-Sequenzierung Technologie, die von PacBio entwickelt wurde und auf der Single Molecule, Real-Time (SMRT) Technologie basiert. Im Gegensatz zu fragmentierten RNA-Sequenzierung Methoden, die eine Rekonstruktion erfordern, sequenzieren mit Iso-Seq direkt vollständige cDNA-Transkripte, die die gesamte 5' untranslatierte Region (5' UTR) bis hin zum 3' Polyadenylierungsschwanz (polyA) umfassen. Dieser Ansatz ermöglicht eine genauere Analyse des Zielgensatzes oder des gesamten Transkriptoms und liefert Einblicke, die auf herkömmliche Weise schwer zu gewinnen sind. Next-Generation Sequencing (NGS) Techniken.
Die Einführung von Iso-Seq
In eukaryotischen Organismen kann ein einzelnes Gen eine überraschend hohe Anzahl von Proteinen kodieren, die nach alternativem Spleißen entstehen, wobei jedes eine distinct biologische Funktion hat. Es gibt bekannte menschliche Gene, die je nach exprimierter Spleißvariante sehr unterschiedliche Funktionen haben. Da alternatives Spleißen so entscheidend für die Funktion des Genoms ist.
Kurzzeit-RNA-Seq arbeitet, indem sie Transkript-Isoformen physisch in kleinere Stücke schneidet und wieder zusammensetzt, was Möglichkeiten für Fehlzusammenstellungen oder unvollständige Erfassung der gesamten Vielfalt von Isoformen aus den interessierenden Genen lässt. Es ist wichtig, vollständige Transkripte zu erfassen. Indem man die Vorteile von PacBio SMRT-Langlesen-Sequenzierung Die Isoform-Sequenzierung (Iso-Seq) kann mithilfe der Technologie leicht vollständige Transkripte vom 5'-Ende bis zum 3'-Poly-A-Schwanz abdecken, ohne dass eine Fragmentierung erforderlich ist, um vollständige cDNA-Sequenzen zu erhalten. Dies ist nützlich, um neue Transkripte und neue Introns zu identifizieren und somit Isoformen, alternative Spleißstellen, die Expression von Fusionsgenen und allelische Expression genau zu bestimmen.
Im Vergleich zur Illumina-Plattform kann die PacBio-Analyse sehr lange polycistronische RNA-Moleküle (sogenannte komplexe Transkripte) leicht erkennen und eine Vielzahl neuartiger transkriptioneller Überlappungen zwischen benachbarten und entfernten Genen, die parallel angeordnet sind, aufdecken. Dies eröffnet die Möglichkeit, ein genomweites Netzwerk zu untersuchen, das eine gemeinsame Kontrolle über die Genexpression und Replikation ausübt.
Wir bieten auch an Nanopore Voll-Längen-Transkript-Sequenzierung Dienstleistungen. Für weitere Informationen besuchen Sie bitte die entsprechende Seite.
Hauptmerkmale und Vorteile von Iso-Seq
- Direkte Erfassung von Volllängen-Transkriptsequenzen, die eine genauere Abbildung der Transkriptom-Informationen für die sequenzierten Arten ermöglichen.
- Erkennung mehrerer alternativer Spleißformen, Aufdeckung zusätzlicher Spleißstellen und alternativer Spleißereignisse.
- Entdeckung neuartiger funktioneller Gene zur Erweiterung der Genomannotation.
- Erleichterung der präzisen Analyse von Fusionsgenen, homologen Genen, Gen-Superfamilien oder Allelen.
- Hohe Genauigkeit, Kontinuität und Vollständigkeit der Transkriptsequenzen.
- Integriert mit der zweiten Generation Transkriptom-Sequenzierung, was eine präzise Quantifizierung auf sowohl Gen- als auch Transkriptebene ermöglicht.
- Hohe Artenanpassungsfähigkeit, die eine Analyse mit oder ohne Referenzgenom ermöglicht.
Anwendungen von Iso-Seq
- Genomische Annotation: Führen Sie eine präzise Annotation neuer Genome durch und liefern Sie hochwertige Transkriptinformationen.
- Transkriptomanalyse: Untersuchen Sie die Vielfalt der Transkripte in verschiedenen Geweben oder Zelltypen und erläutern Sie die Komplexität der Genexpression.
- Krankheitsforschung: Identifizierung spezifischer Genfusionen und alternativer Spleißereignisse in Krebs- und genetischen Krankheitsstudien.
- Agrarbiotechnologie: Anwendung in der Pflanzengenomforschung zur Verbesserung von Zuchtstrategien für verbesserte Pflanzenvarietäten.
Iso-Seq-Workflow
CD Genomics nutzt die PacBio SMRT-Technologie für die Voll-Längen-Transkription-Sequenzierung (Iso-Seq). Diese fortschrittliche Methode umfasst die Extraktion von hochqualitativem RNA, die Synthese von Voll-Längen cDNA, den Aufbau von cDNA-Bibliotheken und die Durchführung von hochpräzisen Sequenzierungen, um vollständige Transkriptsequenzen zu erfassen und eine umfassende und genaue Transkriptomanalyse zu gewährleisten.

Dienstspezifikationen
Beispielanforderungen
|
|
 Klicken |
Sequenzierungsstrategie
|
 |
Bioinformatikanalyse
Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an: Transkriptom ohne Referenz Standarddatenanalyse
Standarddatenanalyse
|
Analyse-Pipeline
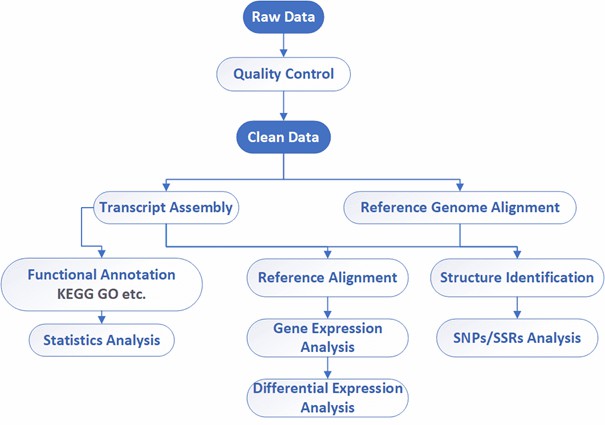
Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details in Iso-Seq für Ihr Schreiben (Anpassung)
Langzeit-Sequenzierung ermöglicht die unkomplizierte Identifizierung von alternativ transkribierten oder bearbeiteten Transkripten, polycistronischen Transkriptionseinheiten und anderen langen cDNA-Sequenzen. Mit Fachwissen und Engagement wird die fortschrittliche PacBio SMRT-Plattform und die Dienstleistungen von CD Genomics Ihr bester Begleiter in der Iso-Seq sein. Bitte kontaktieren Sie uns für weitere Informationen und ein detailliertes Angebot.
Demo-Ergebnisse
Teilweise Ergebnisse sind unten aufgeführt:
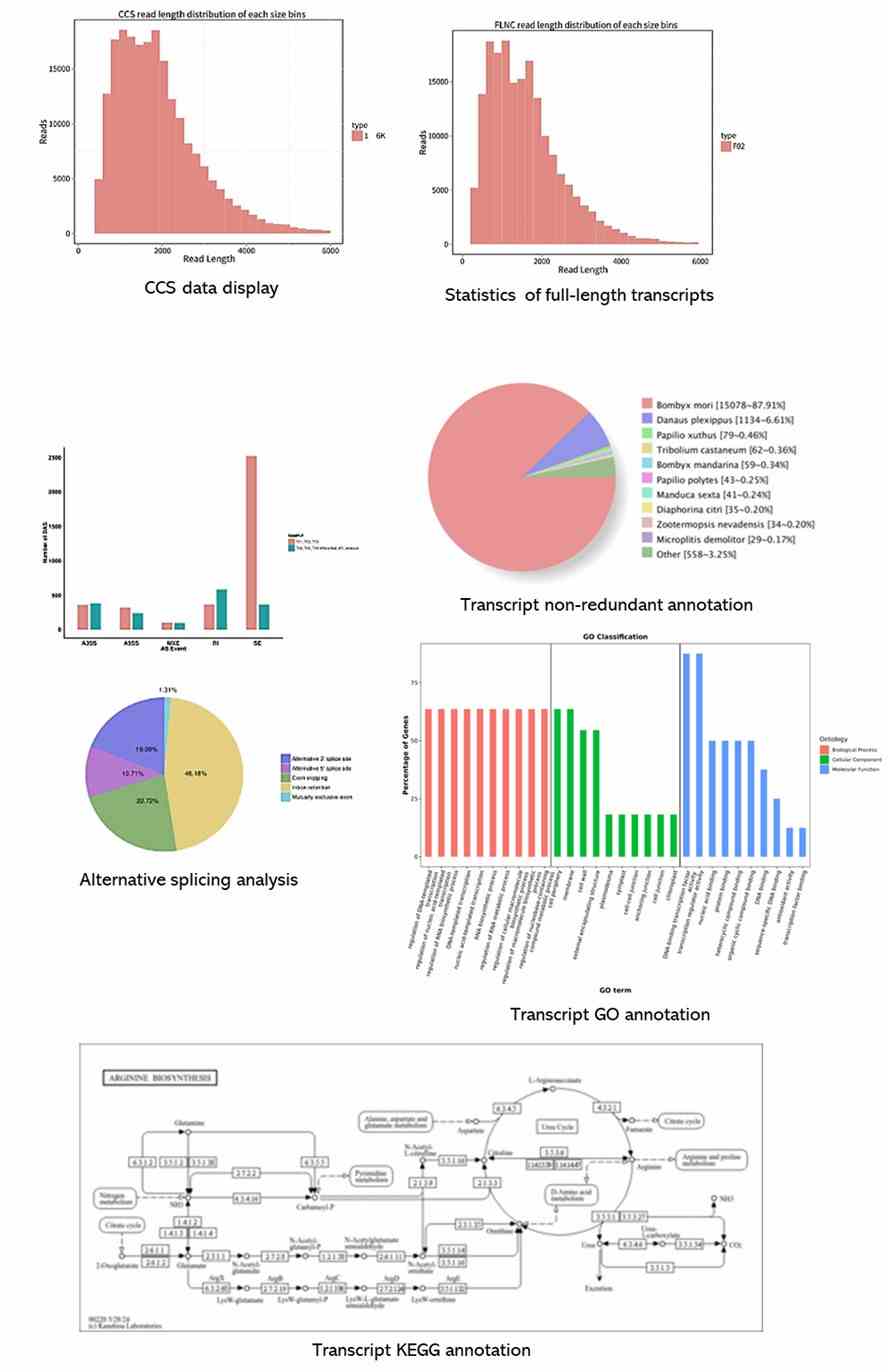
Iso-Seq häufig gestellte Fragen
Im Bereich der Transkriptomik beziehen sich Isoform und Transkript auf unterschiedliche Konzepte. Ein Transkript ist eine RNA-Kopie eines Gens, die während der Transkription erzeugt wird. Eine Isoform hingegen ist eine spezifische Variante eines Transkripts, die durch alternatives Spleißen oder andere posttranskriptionale Modifikationen entsteht. Der Hauptunterschied besteht darin, dass ein Transkript die gesamte RNA-Sequenz eines Gens beschreibt, während eine Isoform eine spezifische Version dieser Sequenz darstellt, die unterschiedliche Eigenschaften oder Funktionen haben kann.
Ein Isoform bezeichnet RNA-Moleküle, die von demselben Genlocus transkribiert werden, aber strukturelle Variationen aufweisen. Im Gegensatz dazu ist ein RNA-Transkript eine RNA-Molekülkopie, die während der Gen-Transkription produziert wird. Im Wesentlichen beschreiben beide Begriffe dasselbe Wesen – RNA-Moleküle. Der Schwerpunkt liegt jedoch unterschiedlich: Isoform hebt strukturelle Unterschiede in RNA-Molekülen hervor, wie TSS, CDS und UTR-Regionen, während Transkript sich auf die posttranskriptionale Verarbeitung bezieht, die die RNA-Moleküle durchlaufen haben, wie zum Beispiel welche Exons miteinander verbunden wurden.
2. Was sind die Eigenschaften von Transkripten, die durch Iso-Seq-Sequenzierung angestrebt werden?
- Typische Struktur: Eukaryotische mRNA-Transkripte bestehen normalerweise aus einer 5'-Kappe, 5'-UTR, CDS, 3'-UTR und einem PolyA-Schwanz. Iso-Seq nutzt PolyA oder spezifische Sequenzen, um mRNA anzureichern oder spezifische Gen-RNAs zu zielen.
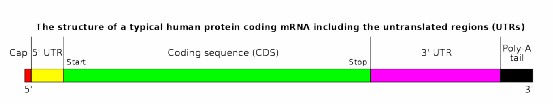 Abbildung 1 Typische mRNA-Struktur.
Abbildung 1 Typische mRNA-Struktur.
- Längenverteilung: Die Transkriptlängen liegen überwiegend zwischen 1 und 3 Kilobasen. Zum Beispiel sind über 99 % der menschlichen Transkripte kürzer als 10 Kilobasen, was es Iso-Seq ermöglicht, die Volllängensequenzierung problemlos zu erreichen.
- Komplexität: Nach der Transkription kann ein Gen alternatives Splicing durchlaufen, um mehrere Transkripte mit unterschiedlichen biologischen Funktionen zu erzeugen. Die genaue Unterscheidung dieser feinen strukturellen Unterschiede zwischen Transkripten hat bedeutende Herausforderungen für Techniken dargestellt, die dem Iso-Seq vorausgingen.
3. Welche Arten von SMRTbell-Designs gibt es für von PacBio unterstützte Iso-Seq-Bibliotheken?
Derzeit umfassen die von PacBio unterstützten Iso-Seq-Sequenzierungsbibliotheken mehrere Arten von SMRTbell-Designs:
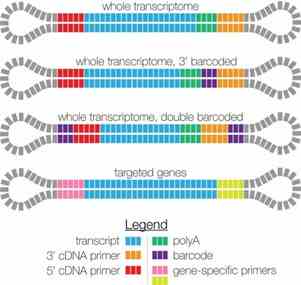 Abbildung 2 SMRTbell-Design.
Abbildung 2 SMRTbell-Design.
Diese Designs ermöglichen flexible Anwendungen: Durch die optionale Hinzufügung von Barcodes unterstützt Iso-Seq sowohl die Einzelproben- als auch die multiplexe Sequenzierung; durch die Verwendung unterschiedlicher Primer für die reversen Transkription erleichtert es sowohl die Ganztranskriptomsequenzierung als auch die gezielte Sequenzierung spezifischer Gen-Sets.
4. Welche grundlegenden Schritte sind an der Datenvorverarbeitung von Iso-seq beteiligt und was sind deren Ziele?
Die Datenvorverarbeitung von Iso-Seq erfolgt durch die Komponenten ccs, lima und IsoSeq3 der SMRT-Tools. Die Schritte umfassen:
- Korrektur zufälliger Sequenzierungsfehler in Subreads im CCS-Sequenzierungsmodus, um hochwertige HiFi-Reads zu erhalten.
- (Bei multiplexen Sequenzierungen) Proben mit Barcodes aufteilen und Barcode-Primer-Sequenzen entfernen.
- Identifizierung und Entfernung von PolyA-Schwänzen und Kettenmolekülen.
- Clustern und Duplikate von Reads entfernen, um Konsenssequenzen zu erzeugen.
Iso-Seq Fallstudien
Nutzung von PacBio Iso-Seq zur Entdeckung neuer Transkripte und Gene in Bezug auf Reaktionen auf abiotischen Stress in Oryza sativa L..
Zeitschrift: Internationale Zeitschrift für Molekulare Wissenschaften
Impactfaktor: 6,208
Veröffentlicht: 31. Oktober 2020
Hintergrund
Der globale Klimawandel verschärft abiotische Stressbedingungen und wirkt sich negativ auf die Ernteerträge aus. Reis, ein Grundnahrungsmittel für über die Hälfte der Weltbevölkerung, weist eine umfangreiche genetische Vielfalt auf. Aktuelle Studien basieren jedoch auf dem Japonica-Referenzgenom, wodurch möglicherweise wichtige genetische Informationen anderer Unterarten verloren gehen. Um dies zu beheben, verwendeten die Autoren PacBio SMRT Iso-Seq, um partielle Transkriptome aus verschiedenen Reissubarten zu sequenzieren und zu rekonstruieren, was die Entdeckung neuer Gene für Stressresistenz ermöglichte, ohne ein vorhandenes Referenzgenom zu benötigen.
Materialien & Methoden
Probenvorbereitung
- Pflanzenmaterial
- Reis
- RNA-Extraktion
Sequenzierung
- RNA-Seq
- De novo Transkriptomrekonstruktion
- PacBio Iso-Seq
- InDel-Analyse
- BUSCO-Analyse
- Phylogenetische Analyse
- Funktionale Annotation
- Differenzielle Genexpressionsanalyse
Ergebnisse
Die Autoren wählten zehn Reissorten aus und isolierten RNA von Pflanzen, die unter verschiedenen Bedingungen angebaut wurden. Die Sequenzierung auf der PacBio-Plattform ergab 15,49 bis 24,51 Gigabasen pro Sorte. Mit IsoSeq3 erhielten sie nach der Filterung von pflanzenspezifischen Sequenzen und der Entfernung von Kontaminanten 37.535 bis 54.594 hochqualitative Voll-Längen-Transkripte pro Sorte.
Tabelle 1. Probenahme für PacBio-Isoform-Sequenzierung.
![]()
Während der Bibliotheksvorbereitung können degradierte 5'-RNA-Produkte zu redundanten Isoformen führen, die vollständige 5'-Sequenzinformationen vermissen lassen. Drei Methoden - Cogent, cDNA Cupcake und TAMA - wurden getestet, um diese Redundanzen zu reduzieren. Während cDNA Cupcake und TAMA ein Referenzgenom verwenden, rekonstruiert Cogent ein kodierendes Genom zur Reduzierung. Alle Methoden verringerten die Anzahl der Isoformen signifikant, wobei Cogent mehr nicht übereinstimmende Transkripte zeigte. Nach der Reduzierung nahmen die Transkriptlängen zu und die Einzigartigkeit der Isoformen verbesserte sich, insbesondere hatte TAMA den höchsten Anteil an einzigartigen Isoformen pro Genlocus. Die phylogenetische Analyse basierend auf SNPs hob deutliche Cluster nach Unterarten hervor und betonte die genetischen Unterschiede zwischen Aus-, Indica- und Japonica-Kultivaren.
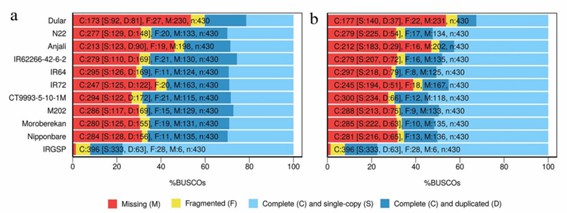 Abbildung 1. BUSCO-Bewertungsanalyse von nicht zusammengefassten (a) und zusammengefassten (b) Transkripten.
Abbildung 1. BUSCO-Bewertungsanalyse von nicht zusammengefassten (a) und zusammengefassten (b) Transkripten.
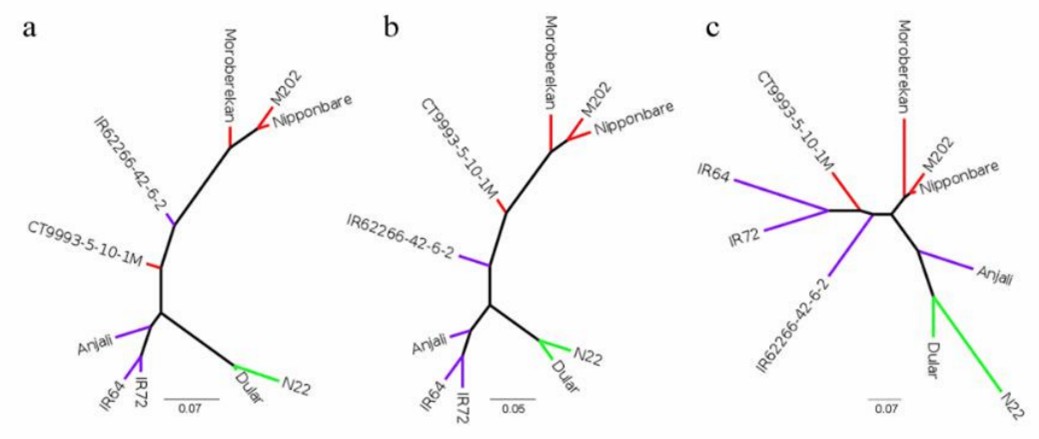 Abbildung 2. Phylogenetische Bäume, die mit SNPhylo erstellt wurden.
Abbildung 2. Phylogenetische Bäume, die mit SNPhylo erstellt wurden.
Die Studie verwendete TAMA, um HQ-Transkripte zusammenzufassen und für nicht zugeordnete Transkripte zu konsolidieren, was zu 10.511 bis 15.011 Genloci und 14.255 bis 20.803 einzigartigen Isoformmodellen pro Sorte führte. Etwa ein Drittel der Genloci und die Hälfte der Transkriptmodelle stimmten mit dem Nipponbare-Referenzgenom überein. Die funktionale Annotation ergab 60 % bis 70 % vollständige ORFs und hob wichtige biologische Prozesse hervor. Ein bemerkenswerter Teil der Transkripte blieb jedoch unannotiert, möglicherweise aus wilden Oryza-Arten stammend.
 Abbildung 3. Anteil der vorhergesagten offenen Leserahmen (ORFs) unter Verwendung von TransDecoder.
Abbildung 3. Anteil der vorhergesagten offenen Leserahmen (ORFs) unter Verwendung von TransDecoder.
Um gemeinsame und spezifische Transkripte unter den Sorten zu identifizieren, wurde eine Sorte aus jeder Unterart (N22, IR64, Nipponbare) als Blast-Datenbank verwendet. Etwa 9.000 Transkripte waren in allen Sorten gemeinsam, wobei 652 einzigartig für N22 und 2426 für IR64 waren. Die Analyse der differentiellen Genexpression in N22 ergab 56 aus-spezifische Gene, die auf kombinierte Trockenheits- und Hitzestressbedingungen ansprechen, einschließlich des hochregulierten B12288-Gens, das Homologie zu trockenheitsreaktiven Genen in anderen Oryza-Arten und Arabidopsis thaliana aufweist.
 Abbildung 4. Multiple Sequenzalignment von fünf Oryza RAB21 Dehydrin-Proteine.
Abbildung 4. Multiple Sequenzalignment von fünf Oryza RAB21 Dehydrin-Proteine.
Fazit
Die Autoren sequenzierten die Transkriptome von zehn Reissorten mit PacBio Sequel und erzielten 37.500 bis 54.600 hochqualitative Isoformen pro Sorte. Ihre de novo Rekonstruktionen umfassten etwa 40 % neuartige Isoformen im Vergleich zu Nipponbare. Für die trockenheits- und hitzebeständige Aus-Sorte N22 identifizierten sie 56 unterschiedlich exprimierte Gene in sich entwickelnden Samen unter Feldbedingungen, was eine kostengünstige Möglichkeit bietet, Gene für Stressresistenz zu identifizieren, die in standardmäßigen Genomassemblierungen fehlen.
Referenz
- Schaarschmidt S, Fischer A, Lawas LM, et al. Nutzung von PacBio Iso-Seq zur Entdeckung neuer Transkripte und Gene in Bezug auf Reaktionen auf abiotischen Stress in Oryza sativa L.. Internationale Zeitschrift für Molekulare Wissenschaften2020, 21(21):8148.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Bakterielle Gemeinschaften von Cassiopea in den Florida Keys teilen sich wichtige bakterielle Taxa mit Korallen-Mikrobiomen.
Journal: bioRxiv
Jahr: 2024
Produktion eines Bakteriocin-ähnlichen Proteins PEG 446 aus Clostridium tyrobutyricum NRRL B-67062
Journal: Probiotika und antimikrobielle Proteine
Jahr: 2024
Entwirrung der Rolle von Pathobionten aus Bacteroides-Arten bei entzündlichen Darmerkrankungen
Journal: bioRxiv
Jahr: 2023
Eine Chromosomenebene-Genomressource zum Studium der Virulenzmechanismen und der Evolution des Kaffeerostpathogens Hemileia vastatrix
Journal: bioRxiv
Jahr: 2022
Streptomyces buecherae sp. nov., ein Actinomycet isoliert aus mehreren Fledermausarten
Journal: Antonie van Leeuwenhoek
Jahr: 2020
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


