
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Bakterielle RNA-Sequenzierung
Neben Eukaryoten RNA-Sequenzierung Der CD Genomics-Service ist auch darauf spezialisiert, prokaryotische RNA-Sequenzierungsdienste anzubieten, um Ihre Bedürfnisse in der Profilierung der bakteriellen Genexpression durch die Nutzung der neuesten Techniken voranzutreiben.
Die Einführung der bakteriellen RNA-Sequenzierung
In den letzten Jahren, Hochdurchsatz-Sequenzierung von cDNA-Bibliotheken (RNA-Seq) hat sich als eine leistungsstarke Technologie zur Profilierung der Genexpression, zur Entdeckung zuvor nicht annotierter Gene und zur Kartierung der Transkriptomarchitektur in einer Vielzahl von Bakterienarten etabliert. Mit der Anwendung von RNA-Seq abgeleitete Ansätze können uns biologische Einblicke in die bakterielle Welt ermöglichen und uns dazu anregen, die Geheimnisse der Genexpression, Organisation und anderer funktioneller genomischer Merkmale zu entschlüsseln. Das bakterielle Transkriptom und Metatranskriptom Information sind wichtig für die Vorhersage von Resistenzen gegen spezifische Antibiotika, das Verständnis der Immuninteraktionen zwischen Wirt und Pathogen, die Quantifizierung von Veränderungen der Genexpression und das Verfolgen des Krankheitsverlaufs.
Es gibt offensichtliche Unterschiede in der Konstruktion von cDNA-Bibliotheken zwischen bakterieller RNA-Seq und eukaryotischer RNA-Seq. Der erste Schritt im Workflow der bakteriellen RNA-Seq ist die Auswahl von mRNA-Transkripten. Die Ribo-Zero-Ribosomen-RNA-Reduktionschemie wird anstelle einer Poly-A-Schwanzauswahl verwendet, was diese Methode besonders geeignet für Bakterien macht, da deren mRNA keinen Poly-A-Schwanz aufweist. Nach der Reinigung wird die mRNA in kleine Stücke fragmentiert und in den ersten Strang cDNA unter Verwendung von reverser Transkriptase und zufälligen Primern kopiert. Die Strangspezifität wird erreicht, indem dTTP im Second Strand Marking Mix (SMM) durch dUTP ersetzt wird, gefolgt von der Synthese des zweiten Strangs cDNA. Durch die Verwendung von strangspezifischer RNA-Seq könnte ein umfassenderes Verständnis des Transkriptoms erreicht werden, was das Potenzial hat, neue Ebenen der Regulation der Genexpression zu identifizieren.
Vorteile der bakteriellen RNA-Sequenzierung
- Genauige Genstruktur-Annotierung: Transkriptom-Sequenzierung In Prokaryoten können kleine Peptide identifiziert werden, die bei der Genomanalyse möglicherweise übersehen werden, was genauere Informationen über die Genstruktur liefert.
- Erkennung und Annotation von nicht-kodierenden regulatorischen Regionen der mRNA: Die Transkriptom-Sequenzierung offenbart wichtige regulatorische Elemente in prokaryotischer mRNA, wie Riboswitches und Bindungsstellen für kleine RNAs, und verbessert unser Verständnis der regulatorischen Mechanismen von mRNA.
- Studie der Operonstruktur und -funktion: Die Transkriptomanalyse kann Operonstrukturen bestimmen und unser Verständnis der multifunktionalen Regulation und Evolution von polycistronischen Transkripten voranbringen.
- Forschung zur Regulation von nicht-kodierenden RNAs: Durch die Sequenzierung von prokaryotischen sRNAs gewinnen wir Einblicke in die Funktion und Mechanismen dieser regulatorischen Elemente in der physiologischen Kontrolle.
- Plastizität der Funktionen von RNA-Elementen: Die Transkriptom-Sequenzierung zeigt die funktionale Vielseitigkeit von RNA-Elementen unter verschiedenen Bedingungen und trägt zum Verständnis ihrer Rollen in unterschiedlichen Umgebungen bei.
Anwendung der bakteriellen RNA-Sequenzierung
- Untersuchung der molekularen Regulationsmechanismen von Mikroorganismen in Bezug auf verschiedene Stadien, Phänotypen und Stressreaktionen.
- Verbesserung der mikrobiellen Transkriptominformationen, einschließlich der Bestimmung der Transkriptstruktur, der Vorhersage neuer Transkripte und der Identifizierung nicht-kodierender RNAs.
- Genexpressionsebene
- Genstruktur-Ebene
- Genfunktion
- Interaktionsnetzwerk
- Identifikation von differentiellen Genen
- Erkennung von nicht-kodierenden RNAs usw.
Bakterielle RNA-Sequenzierungs-Workflow
Die primären Phasen der bakteriellen RNA-Sequenzierung beginnen mit der Extraktion von Gesamt-RNA aus Bakterienzellen. Dieser Vorgang wird gefolgt von der Eliminierung von rRNA, die die Gesamt-RNA dominiert, um die mRNA-Population selektiv zu erhöhen. Anschließend wird die mRNA durch reversen Transkription in komplementäre DNA (cDNA) umgewandelt. Die resultierende cDNA wird fragmentiert, Adapter werden ligiert und eine PCR-Amplifikation wird durchgeführt, um die Sequenzierungsbibliothek zu erstellen. Anschließend wird eine Hochdurchsatz-Sequenzierung unter Verwendung von Plattformen wie Illumina oder PacBio durchgeführt. Die abschließende Phase umfasst einen sorgfältigen Datenanalyseprozess, der Qualitätssicherung, Sequenzalignment, Quantifizierung und die Untersuchung der differentiellen Expression umfasst.

Dienstspezifikationen
Musteranforderungen
|
|
 Klicken |
Sequenzierungsstrategie
|
 |
Bioinformatikanalyse Analyse auf der Ebene der Genstruktur
|
Analyse-Pipeline

Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details zur bakteriellen RNA-Sequenzierung für Ihre Schreibanpassung.
CD Genomics bietet eine schnelle, umfassende Lösung für die bakterielle RNA-Sequenzierung, die von der Qualitätskontrolle der Proben bis zur umfassenden Datenanalyse reicht. Kontaktieren Sie uns für weitere Informationen und ein detailliertes Angebot.
Referenzen
- Kröger C, MacKenzie K D, Alshabib E Y, et al. Das primäre Transkriptom, kleine RNAs und die Regulation der antimikrobiellen Resistenz in Acinetobacter baumannii ATCC 17978. Nukleinsäureforschung, 2018, 46(18): 9684-9698.
- Liu X, Shen B, Du P, et al. Transkriptomische Analyse der Reaktion von Pseudomonas fluorescens auf Epigallocatechingallat mittels RNA-Seq. PloS One, 2017, 12(5): e0177938.
Demonstrationsergebnisse
Teilweise Ergebnisse sind unten aufgeführt:

Sequenzierungsqualitätsverteilung
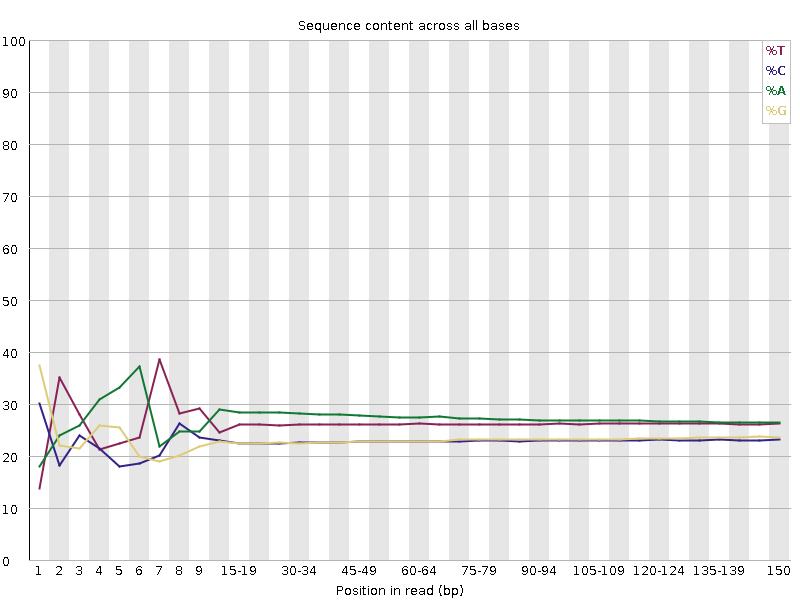
A/T/G/C-Verteilung
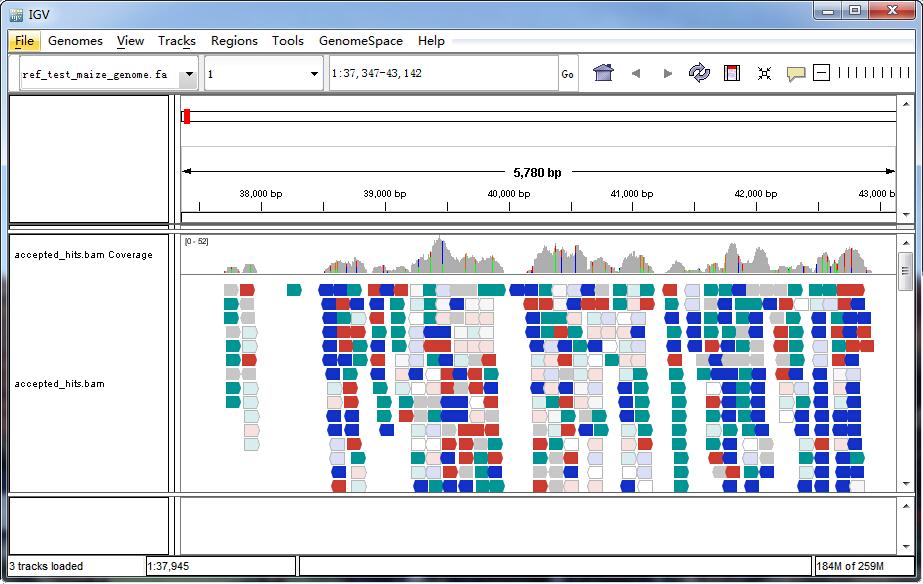
IGV-Browser-Oberfläche
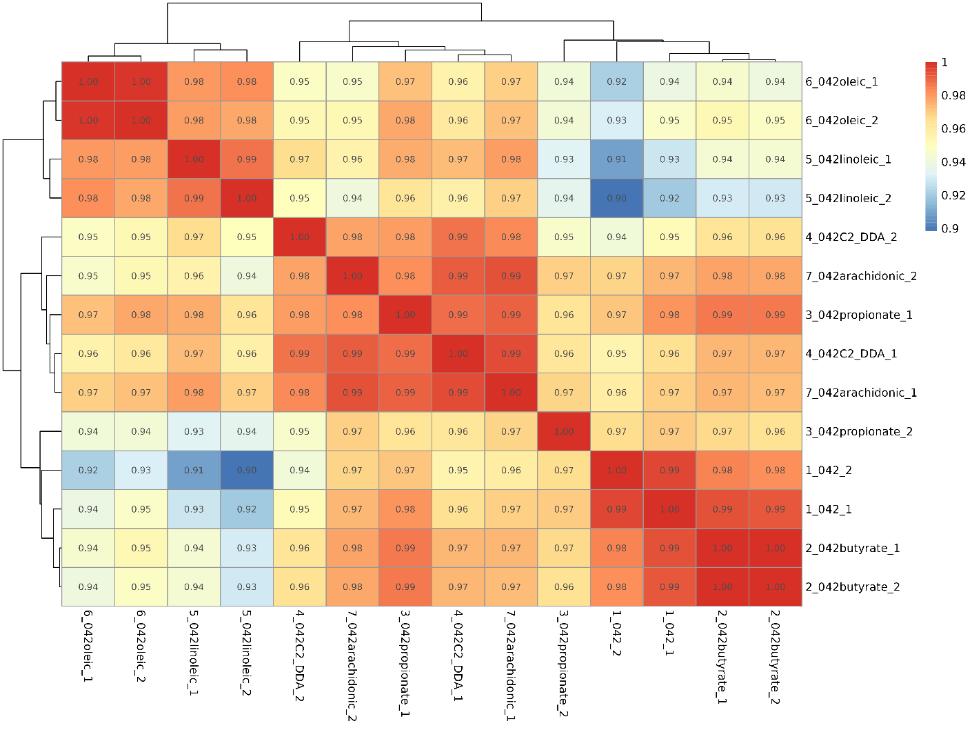
Korrelationsanalyse zwischen Proben
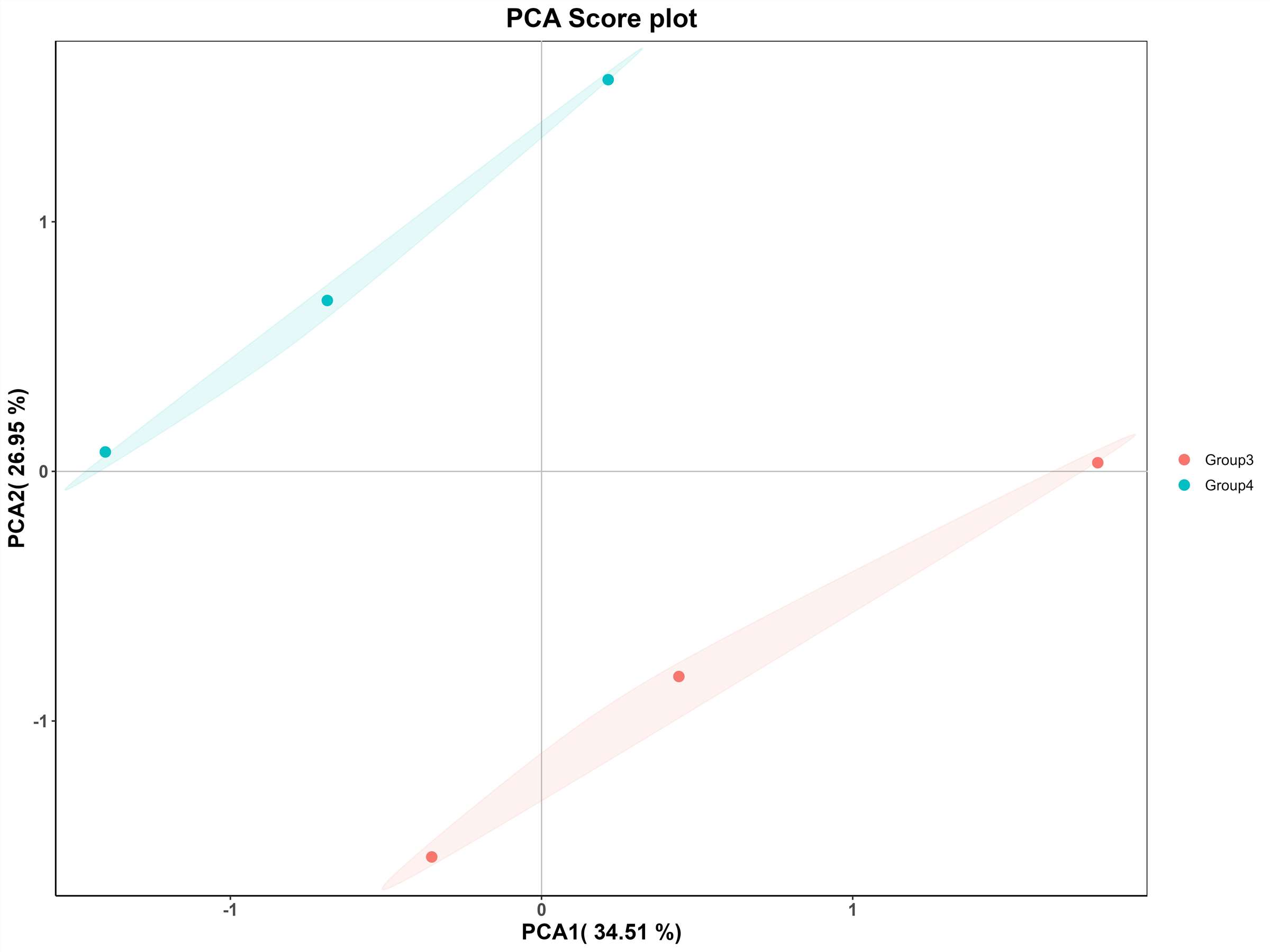
PCA-Score-Diagramm

Venn-Diagramm
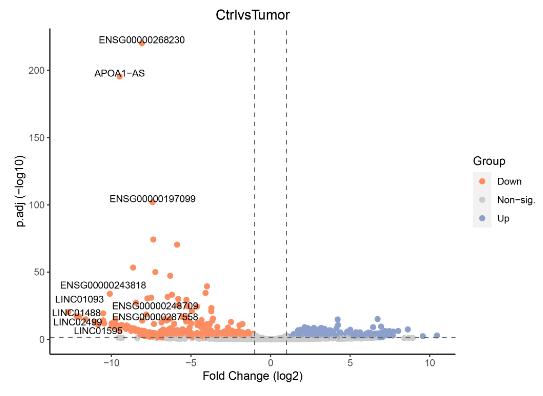
Volcano-Diagramm
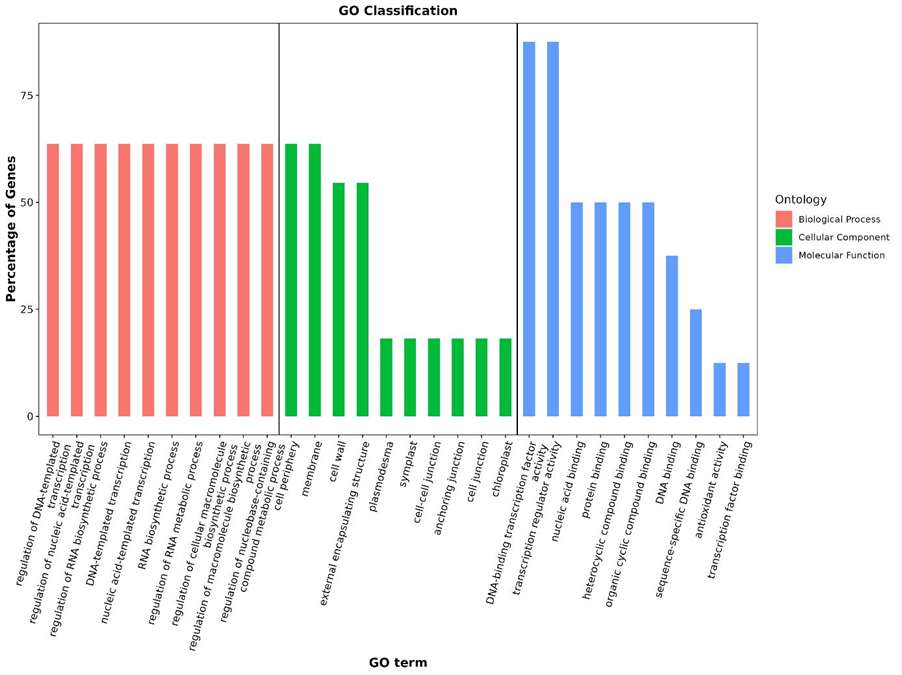
Statistik Ergebnisse der GO-Annotation
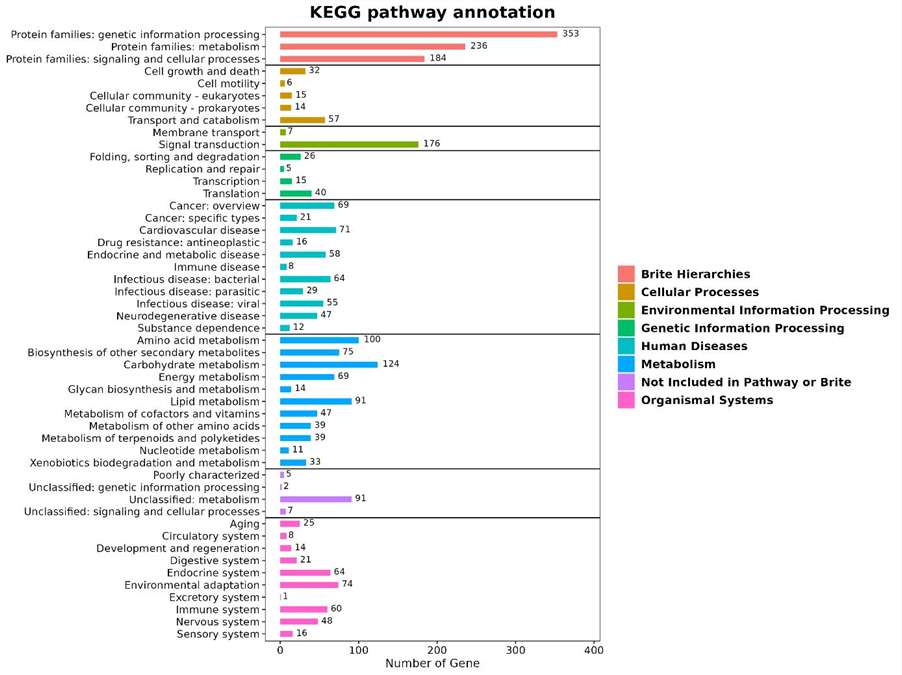
KEGG-Klassifikation
Häufig gestellte Fragen zu bakterieller RNA-Sequenzierung
1. Warum ist die Entfernung von rRNA notwendig?
Die sorgfältige Entfernung von ribosomaler RNA (rRNA) hat eine entscheidende Bedeutung in der bakteriellen RNA-Sequenzierung, da sie in bakteriellen RNA-Proben häufig über 90 % des gesamten RNA-Gehalts ausmacht. Da die messenger RNA (mRNA) – das Hauptziel der Sequenzierungsbemühungen – nur einen geringen Anteil am gesamten RNA-Pool ausmacht, ist eine effektive rRNA-Depletion unerlässlich, um die überwältigende Fülle an rRNA-Sequenzen in Sequenzierungsdatensätzen zu verringern. Das Versäumnis, rRNA zu eliminieren, kann die Sequenzierungsgenauigkeit erheblich gefährden und die Sensitivität der mRNA-Detektion beeinträchtigen.
2. Ist es möglich, die Transkriptom-Sequenzierung von Prokaryoten ohne Referenzgenom durchzuführen?
Nein, da es praktisch nicht machbar ist. Prokaryotische mRNA ist häufig polycistronisch, was eine direkte Assemblierung unzuverlässig macht.
3. Wie unterscheidet sich die Einzelzell-RNA-Sequenzierung von der bakteriellen RNA-Sequenzierung?
Die Einzelzell-RNA-Sequenzierung hebt sich von der bakteriellen RNA-Sequenzierung ab, da sie den Fokus auf die Untersuchung der Genexpression auf der Ebene einzelner Zellen legt und Zell-zu-Zell-Variationen aufdeckt. Im Gegensatz zur bakteriellen RNA-Sequenzierung, die die durchschnittliche Genexpression einer bakteriellen Population untersucht, liegt die Einzigartigkeit der Einzelzell-RNA-Sequenzierung in ihren unterschiedlichen Ansätzen zur Probenvorbereitung, Datenanalyse und potenziellen Anwendungen.
Bakterielle RNA-Seq-Fallstudien
Der Anspruch auf die Vorrangstellung des menschlichen Darms Bacteroides ovatus in der diätetischen Cellobiose-Abbau
Journal: Gut-Mikroben
Impact-Faktor: 11,724
Veröffentlicht: 22. Juni 2023
Hintergrund
Cellulose, ein Hauptbestandteil der Pflanzenzellwände, kann von Säugetieren ohne die Unterstützung von cellulolytischen Bakterien nicht verdaut werden. Diese Bakterien, die im Verdauungstrakt verschiedener Tiere, einschließlich Menschen, leben, spielen eine wesentliche Rolle beim Abbau von Cellulose und liefern dadurch Energie an ihre Wirte. Obwohl eine erhebliche Anzahl cellulolytischer Bakterien identifiziert wurde, sind viele noch unzureichend charakterisiert. Die Schlüsselgene, die für die Produktion von Cellulase verantwortlich sind, finden sich überwiegend in den Glycosid-Hydrolase (GH) Familien. Bei Menschen wurde die Cellulase-Aktivität bemerkenswert beobachtet in Ruminococcus champanellensis, aber weitere Untersuchungen sind unerlässlich, um seine funktionalen Mechanismen vollständig zu erläutern. Jüngste Fortschritte, die unter Verwendung von RNA-Sequenzierung (RNA-Seq) und 16S rRNA-Sequenzierung, konzentrieren sich darauf, neue Cellulasen zu entdecken und die komplexen Prozesse zu verstehen, die dem Abbau von Cellulose im menschlichen Darm zugrunde liegen.
Methoden
Probenvorbereitung:
Lactobacillus rhamnosus GG
Lactobacillus reuteri (LR)
Bifidobacterium longum (BL)
Sequenzierung:
Mikrobielle Analyse
Statistische Analyse
Ergebnisse
Um die PULs zu untersuchen, die für den Zellobioseabbau verantwortlich sind, wurde BO, das mit Zellobiose ko-kultiviert wurde, einer RNA-Sequenzierung unterzogen. Die Ergebnisse zeigten 91 Gene mit veränderter Expression als Reaktion auf Zellobiose, von denen 19 im Vergleich zu BO, das auf Glukose gewachsen war, signifikant hochreguliert waren. Diese hochregulierten Gene wurden in zwei PULs organisiert, PUL1 und PUL2, was durch RT-qPCR bestätigt wurde. PUL1, das aus 15 Genen besteht, darunter Glycosidase-Hydrolasen (GH5 und GH2) und sechs hypothetische Proteine, sowie PUL2, das aus fünf Genen besteht, darunter GH16 und GH3, sind am Zellobioseabbau beteiligt. Bemerkenswert ist, dass PUL1 zuvor nicht berichtet wurde.
 Abbildung 1. Polysaccharid-Nutzungsorte (PULs) wurden durch RNA-seq untersucht und durch RT-qPCR bestätigt.
Abbildung 1. Polysaccharid-Nutzungsorte (PULs) wurden durch RNA-seq untersucht und durch RT-qPCR bestätigt.
Um die spezifischen Wege zu erläutern, durch die der Cellobiose-Abbau im Bakterium erleichtert wird, wurden rekombinante Proteine, die jedes Polysaccharid-Nutzungs-Lokus (PUL) repräsentieren, isoliert und Immunseren zur Charakterisierung produziert. Offensichtlich zeigten die Proteine BACOVA_02626GH5, BACOVA_02630GH5 und BACOVA_02745GH3 eine spezifische Hochregulation in Gegenwart von Cellobiose, was auf ihre direkte Beteiligung am Abbau dieses Substrats hinweist. Durch eine Kombination aus Immunfluoreszenz- und Western-Blot-Analysen wurde festgestellt, dass BACOVA_02630GH5 überwiegend in der Zellmembran lokalisiert ist, während BACOVA_02626GH5 und BACOVA_02745GH3 in relativ niedrigeren Mengen innerhalb der Zelle exprimiert wurden. Enzymatische Bewertungen bestätigten zudem die Fähigkeit von BACOVA_02626GH5, BACOVA_02630GH5 und BACOVA_02745GH3, die Umwandlung von Cellobiose in Glukose zu katalysieren. Diese kollektiven Beobachtungen unterstreichen die unverzichtbare Rolle, die diese Proteine im Cellobiose-Abbauweg innerhalb des Bakteriums spielen.
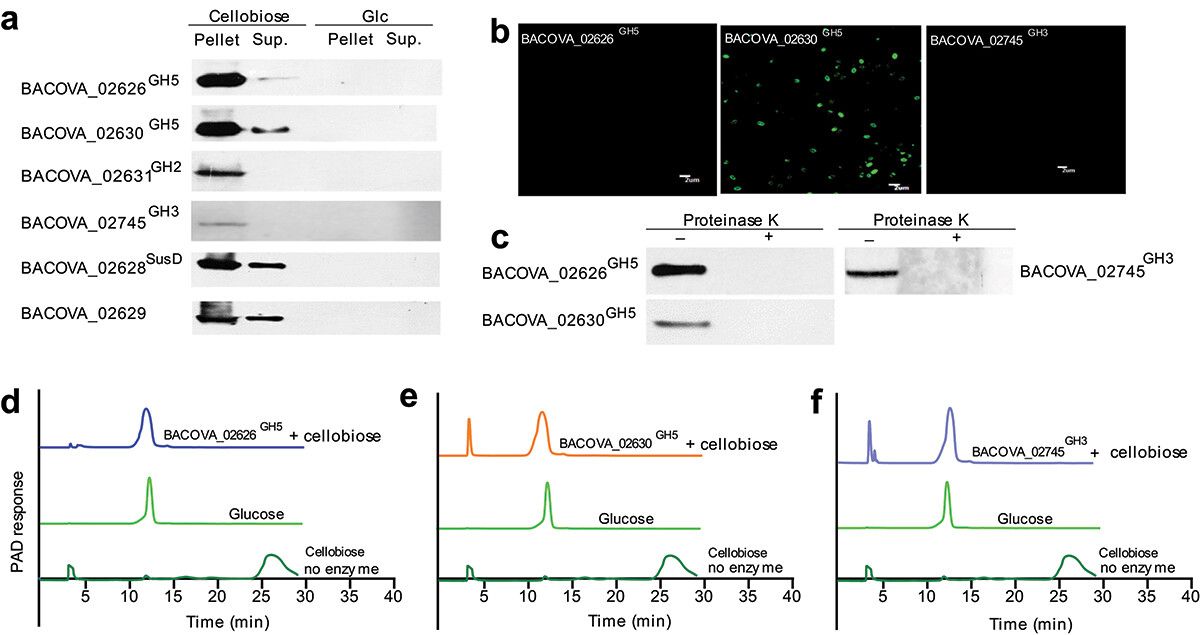 Abbildung 2. Zwei neue Cellulasen wurden bestimmt.
Abbildung 2. Zwei neue Cellulasen wurden bestimmt.
Um bakterielle funktionale Aktivitäten zu untersuchen, verwendeten wir PICRUSt1/2 und Tax4Fun, um 16S rRNA-Sequenzen auf Gene und Stoffwechselwege abzubilden. Die unsupervised Clusteranalyse von 49 vorhergesagten metabolischen MetaCyc-Wegen zeigte signifikante Unterschiede zwischen der Fahrzeug- und der Cellobiose-Gruppe, was auf cellobiose-spezifische mikrobiomale Stoffwechselwege hinweist. Tax4Fun sagte voraus, dass nur β-N-Acetylhexosaminidase, Alpha-Glucosidase und β-Galaktosidase von der Fahrzeuggruppe abwichen. Die PICRUSt1-Analyse ergab angereicherte KEGG-Wege, die mit der Histidin-, Methan-, Vitamin B6- und Folsäurebiosynthese nach der Cellobiose-Behandlung in Verbindung standen. Der Histidin-Stoffwechsel und β-N-Acetylhexosaminidase haben sich als hemmend bei der Kolonentzündung erwiesen. Die Cellobiose-Behandlung verringerte die mRNA- und Proteinspiegel von TNF-α und IL-6 und erhöhte die mRNA-Expression von H2R. Diese Ergebnisse deuten darauf hin, dass Cellobiose die bakteriellen Stoffwechsel-Funktionen verändern könnte, obwohl ihre Auswirkungen auf Entzündungen unklar bleiben und weiterer Untersuchungen bedürfen.
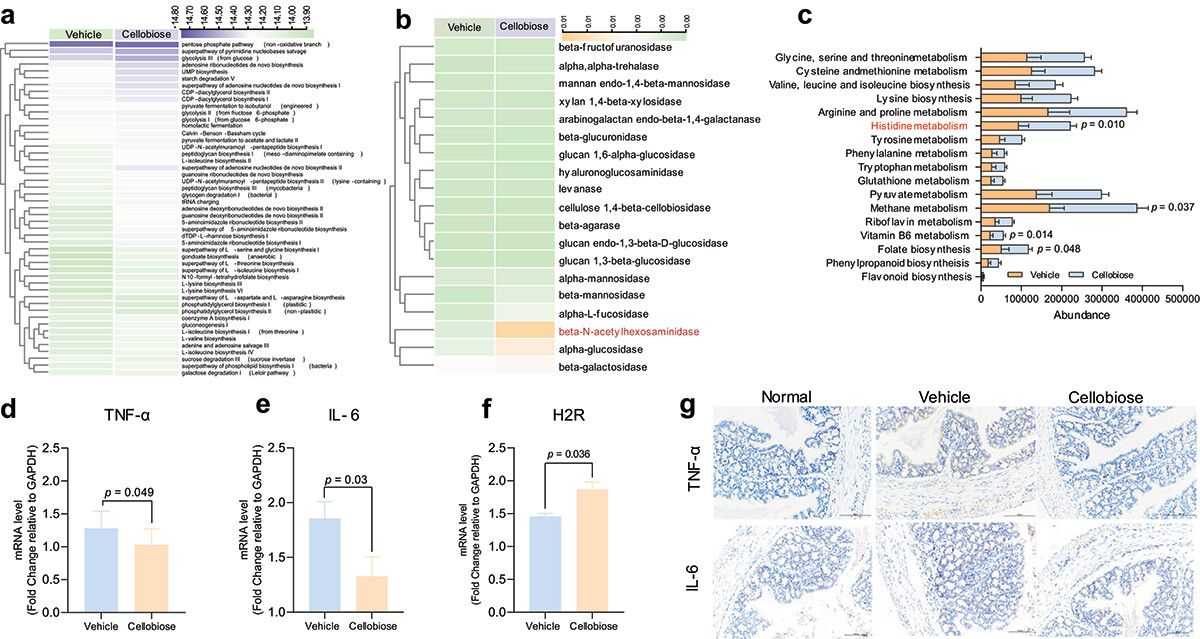 Abbildung 3. Vorhersage der Stoffwechselfunktion von Bakterien und Nachweis von Entzündungsfaktoren.
Abbildung 3. Vorhersage der Stoffwechselfunktion von Bakterien und Nachweis von Entzündungsfaktoren.
Fazit
Cellulolytische Bakterien sind entscheidend für den Abbau von Cellulose, doch ihre molekularen Mechanismen sind noch unzureichend erforscht. Cellobiose, eine Celluloseeinheit, fördert das Wachstum des wichtigen Darmbakteriums BO, aber der molekulare Mechanismus ist unklar. Diese Studie identifizierte zwei neue Zelloberflächen-Cellulasen in BO, die Cellobiose in Glukose abbauen. Diese Cellulasen, deren Strukturen den Enzymen von Bodenbakterien ähneln, deuten auf eine evolutionäre Anpassung hin. In-vivo-Tests zeigten, dass Cellobiose die Darmmikrobiota umgestaltete und Entzündungen reduzierte, was auf potenzielle entzündungshemmende Eigenschaften hindeutet. Diese Forschung erweitert unser Verständnis des Celluloseabbaus im menschlichen Darm und hebt die Notwendigkeit weiterer Erkundungen der cellulolytischen Fähigkeiten der Darmmikrobiota hervor.
Referenz
- Li M, Wang Y, Guo C, et al. Der Anspruch auf die Vorrangstellung von Bacteroides ovatus im menschlichen Darm bei der Abbau von diätetischem Cellobiose. Gute Mikroben, 2023, 15(1): 2227434.
Verwandte Publikationen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Die Spaltung von Phagen-DNA durch Restriktionsendonukleasen ermöglicht die Wiederbelebung aus der durch Cas13 induzierten bakteriellen Dormanz.
Zeitschrift: Nature Mikrobiologie
Jahr: 2023
IL-4 fördert die Erschöpfung von CD8.+ CART-Zellen
Zeitschrift: Nature Communications
Jahr: 2024
Fettreiche Diäten während der Schwangerschaft führen zu Veränderungen der DNA-Methylierung und Proteinexpression im Pankreasgewebe der Nachkommen: Ein Multi-Omics-Ansatz
Internationale Zeitschrift für Molekulare Wissenschaften
Jahr: 2024
KMT2A assoziiert mit dem PHF5A-PHF14-HMG20A-RAI1 Subkomplex in Stammzellen des Pankreaskarzinoms und reguliert epigenetisch deren Eigenschaften.
Zeitschrift: Nature Communications
Jahr: 2023
Krebsassoziierte DNA-Hypermethylierung von Polycomb-Zielen erfordert die duale Erkennung von Histon H2AK119-Ubiquitinierung und der sauren Tasche des Nukleosoms durch DNMT3A.
Journal: Wissenschaftliche Fortschritte
Jahr: 2024
Genomisches Imprinting-ähnliches monoalleles väterliches Ausdrucksmuster bestimmt das Geschlecht von Kanalkatzenfischen.
Journal: Wissenschaftliche Fortschritte
Jahr: 2022
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


