
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Ultra-Niedrig-Eingangs-RNA-Seq (SMART-seq)
Die Einführung der Ultra-Niedrig- RNA-Sequenzierung
Wir bieten einen Ultra-Niedrig-Eingangs-RNA-Sequenzierungsdienst an, der die Untersuchung von Proben mit einer begrenzten Anzahl von Zellen oder mit einer extrem geringen Menge an Eingangs-RNA ermöglicht. Die Ultra-Niedrig-Eingangs-RNA-Sequenzierung ermöglicht es Forschern, die wahre Vielfalt der Genexpression innerhalb kleiner Zellpopulationen in komplexen Geweben zu erforschen und die Reaktionen zellulärer Subpopulationen auf Umweltreize zu verstehen. Sie bietet ein leistungsstarkes neues Werkzeug für die Analyse von außergewöhnlich seltenen oder wertvollen Proben – einschließlich Stammzellen, zirkulierenden Tumorzellen und Hirngewebe-Biopsien.
CD Genomics nutzt die umfassend erprobte Technologie der Branche, um hochqualitatives cDNA mit extrem niedrigen RNA-Spiegeln zu erzeugen, kombiniert mit Illumina-Sequenzierung, um ein leistungsstarkes neues Werkzeug für die Analyse dieser äußerst begrenzten Proben bereitzustellen. Wir bieten mehrere verschiedene Dienstleistungen an, um ein genaues Bild der Expressionsniveaus von kodierenden und nicht-kodierenden RNAs aus kleinen Probenmengen zu erhalten.
Beginnend mit Pikogramm-Mengen von RNA oder wenigen Hundert Zellen bietet die aktuelle Technologie eine unvergleichliche Empfindlichkeit und unvoreingenommene Amplifikation von cDNA-Transkripten, die anreichern für Vollständige Transkripte und bewahrt die wahre Darstellung des Originals mRNA-Transkripte.
Während NGS Die Technologie hat erheblich zu unserem Verständnis von zellulärem mRNA beigetragen, sie hat auch die Existenz einer Vielzahl von nicht-kodierenden RNAs offenbart, die unterschiedliche Rollen in Prozessen wie der Regulierung der Genexpression spielen. Die Ultra-Niedrig-Eingangs-Gesamt-RNA-Sequenzierung ermöglicht die Bereitstellung von Daten, die sowohl mRNA als auch lncRNA umfassen, und ermöglicht die Analyse dieser wichtigen Klasse von regulatorischen RNAs.
Wir bieten auch einen ultra-niedrigen Input-Service für kleine RNAs an, der direkt mit totaler RNA oder angereicherter RNA arbeitet. kleine RNA Eingaben, die von Nanogramm-Mengen an RNA reichen. Es ermöglicht Forschern, eine breite Palette von smRNA-Spezies zu analysieren und Sequenzierungsbibliotheken von erheblicher Komplexität aus nur 1 ng Eingabematerial zu erstellen, wodurch sichergestellt wird, dass verschiedene smRNA-Spezies mit minimalem Bias repräsentiert sind.
Vorteile und Merkmale der Ultra-Niedrig RNA-Sequenzierung
- Beispiel für eine Datenlösung mit der höchsten Datenqualität und den niedrigsten Kosten.
- Genauigkeit der Genquantifizierung und unvergleichliche Empfindlichkeit für ultraniedrigen RNA-Eingang
- Leistungsstarke Analyse von Genexpressionsniveaus und alternativ gespleißten Isoformen, Charakterisierung von kleinen RNAs und Genentdeckung.
- Die ultraniedrig-input RNA-Sequenzierungsmethode zielt darauf ab, Amplifikationsverzerrungen zu minimieren und gleichzeitig die Repräsentation der im ursprünglichen Proben vorhandenen RNA-Spezies zu bewahren. Durch die Optimierung der Schritte der reversen Transkription und der PCR-Amplifikation werden künstliche Artefakte minimiert, was ein umfassendes Verständnis des Transkriptoms gewährleistet.
Ultra-Niedrig RNA-Sequenzierungs-Workflow
Unsere umfassenden Ultra-Niedrig-RNA-Sequenzierungsdienste bieten die RNA-Sequenzierung Workflow von der Probenvorbereitung bis zur Datenanalyse, ermöglicht eine schnelle Profilierung und tiefgehende Einblicke in die RNA.

Dienstspezifikation
Musteranforderungen
|
|
 Klicken |
Sequenzierungsstrategien
|
 |
Datenanalyse Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an:
Hinweis: Die empfohlenen Datenoutputs und Analyseinhalte, die angezeigt werden, dienen nur zur Referenz. Für detaillierte Informationen bitte Kontaktieren Sie uns mit Ihren maßgeschneiderten Anfragen. |
Analyse-Pipeline
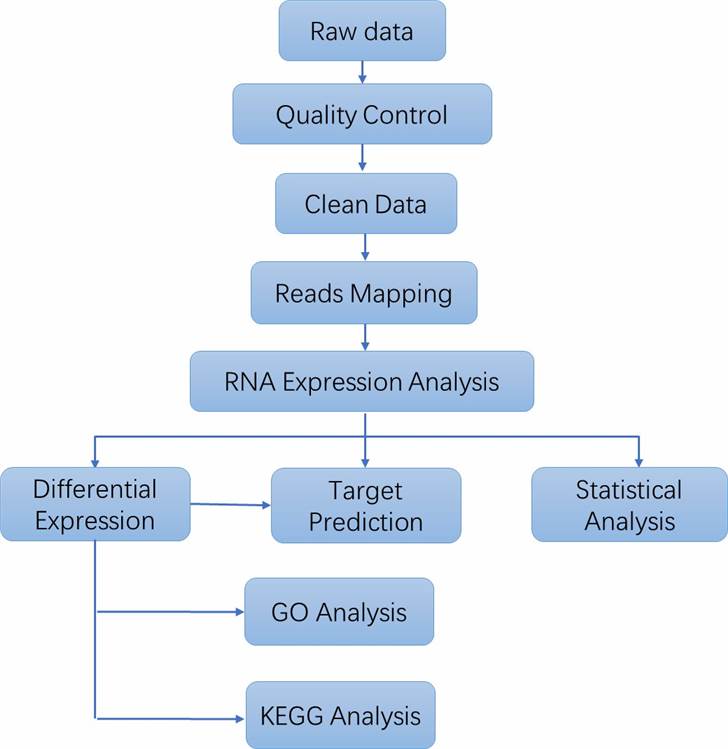
Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details zur Ultra Low RNA-Sequenzierung für Ihre Schreibanpassungen.
Durch den Einsatz unseres Teams erfahrener Experten und modernster Technologie bietet CD Genomics einen spezialisierten Ultra Low RNA Sequencing-Service an. Unsere Plattform liefert hochauflösende RNA-Sequenzdaten durch strenge Qualitätskontrollen und fortschrittliche bioinformatische Analysen, die unvergleichliche Genauigkeit und Sensitivität gewährleisten.
Demonstrationsergebnisse
Teilweise Ergebnisse sind unten aufgeführt:

Sequenzierungsqualitätsverteilung
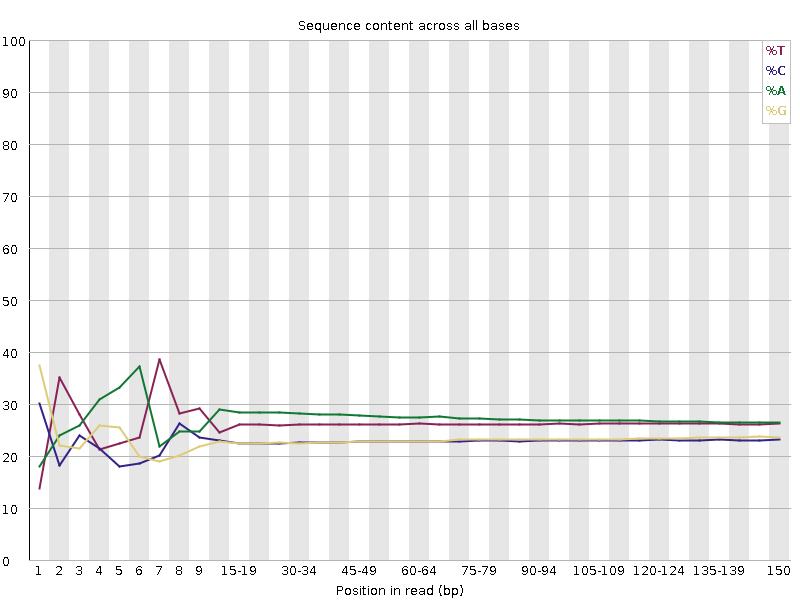
A/T/G/C Verteilung
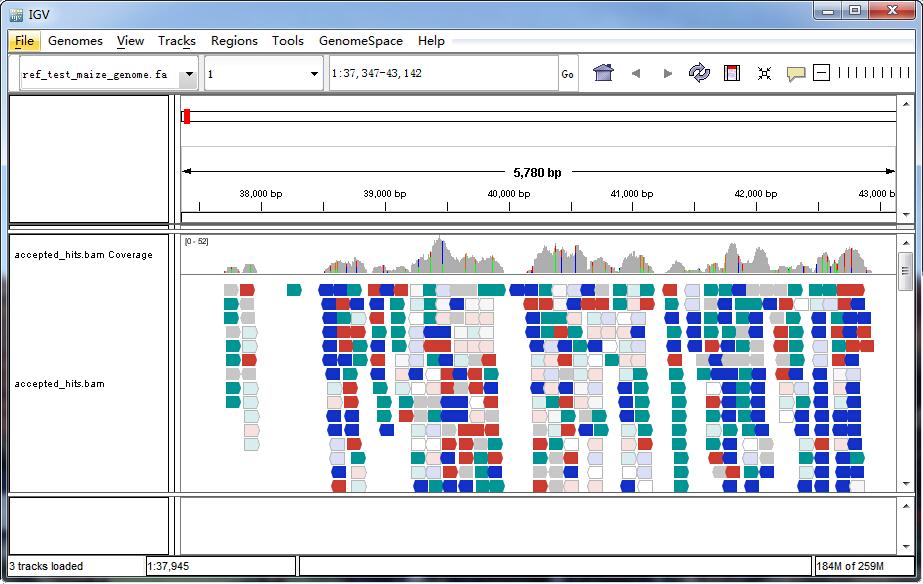
IGV-Browser-Oberfläche
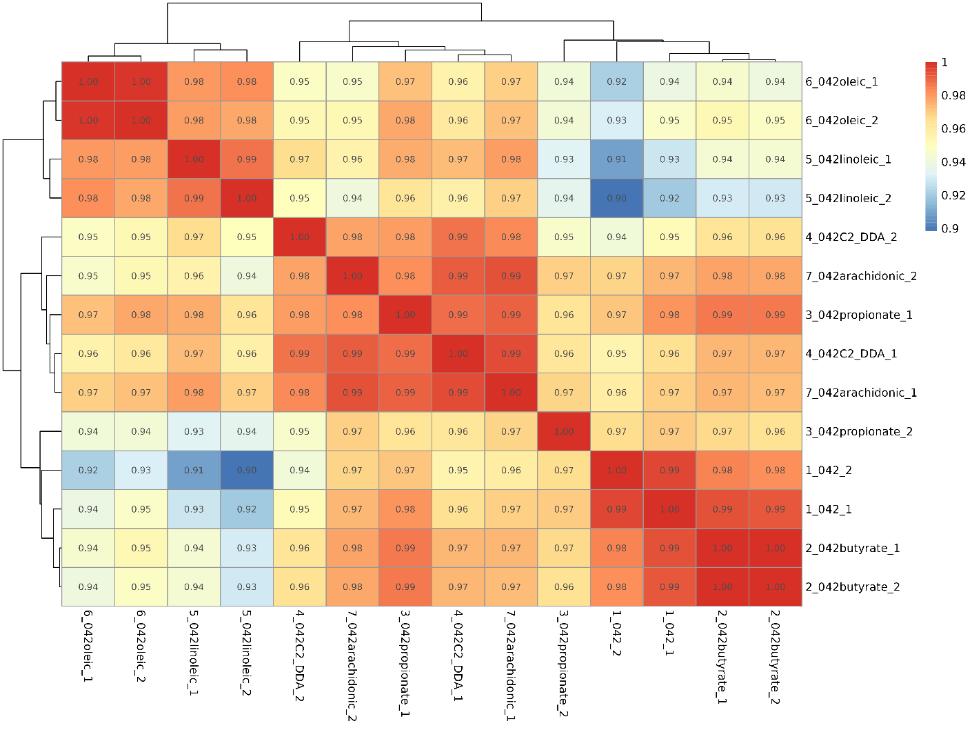
Korrelationsanalyse zwischen Proben
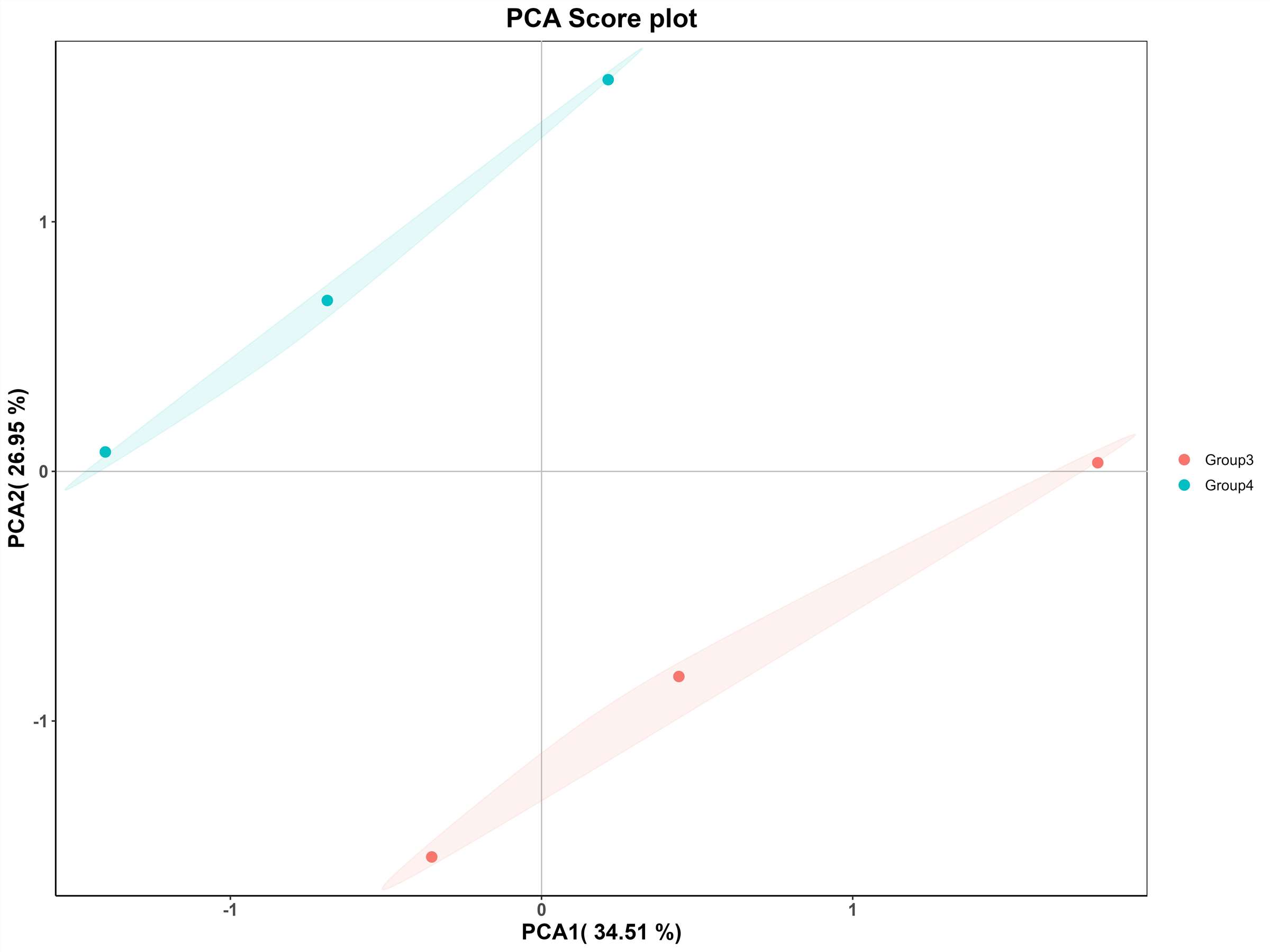
PCA-Score-Diagramm

Venn-Diagramm
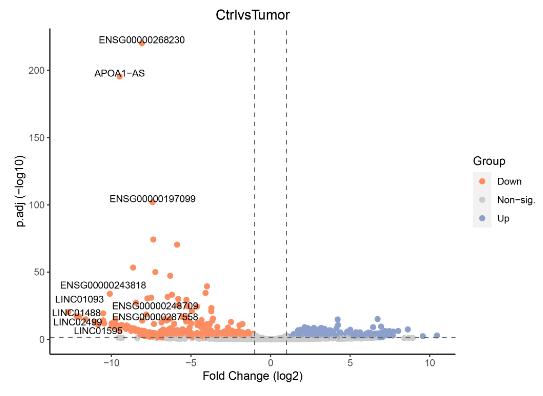
Vulkan-Plot
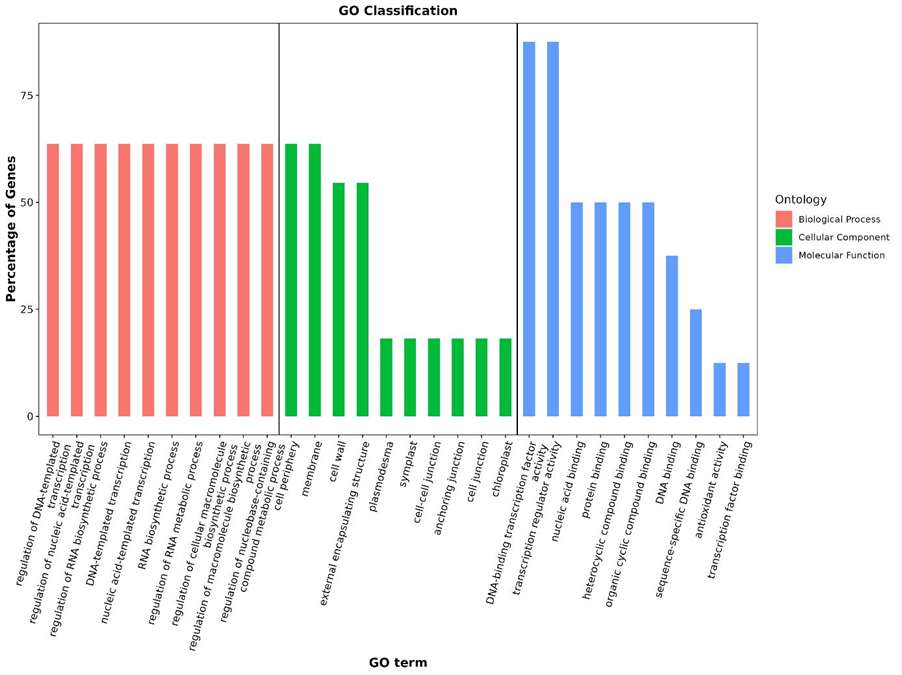
Statistik Ergebnisse der GO-Anmerkung
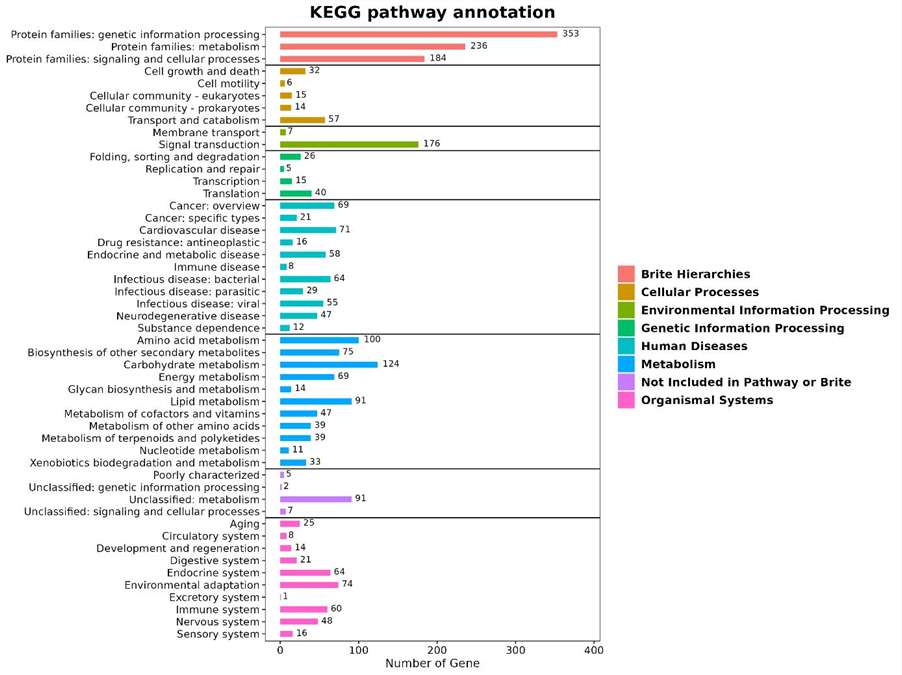
KEGG-Klassifikation
SMART-seq häufige Fragen (FAQs)
Was ist Ultra Low RNA-Sequenzierung?
Ultra Low RNA-Sequenzierung (ULRS) stellt eine hochmoderne Technologie dar, die darauf ausgelegt ist, hochgenaue Sequenzierungsdaten aus winzigen Mengen von RNA-Proben zu erzeugen. Diese Methode ist strategisch positioniert, um komplexe biologische Fragestellungen zu adressieren, wie zum Beispiel die Einzelzell-RNA-Sequenzierung (scRNA-seq), die Analyse seltener Zellpopulationen und das Profiling von RNA, die aus klinischen Proben extrahiert wurde.
2. Was sind die Anwendungen der Ultra Low Input RNA-Sequenzierung?
Ultra-Niedrig-Eingangs-RNA-Sequenzierung ist in mehreren Bereichen von entscheidender Bedeutung: Sie ermöglicht es der scRNA-seq, zelluläre Transkriptome zu untersuchen, identifiziert seltene Zelltypen und extrahiert RNA aus klinischen Proben für diagnostische Einblicke. In der Entwicklungsbiologie und der Stammzellforschung klärt ULRS die Dynamik der Genexpression und bietet nuancierte RNA-Profiling in verschiedenen biologischen Kontexten, wodurch das wissenschaftliche Verständnis vorangetrieben wird.
3. Was sind die Anforderungen an Proben für Ultra Low RNA-Sequenzierung?
Bei der Ultra Low RNA Sequenzierung (ULRS) ist die Integrität der Proben ein entscheidender Faktor, um RNA-Verschlechterung zu vermeiden, was hochwertige RNA-Eingaben erfordert. Die Empfindlichkeit der Methode gegenüber geringen RNA-Mengen hebt die Kompatibilität von Proben im Bereich von Pikogramm bis Nanogramm hervor. Die Aufrechterhaltung der Probenreinheit dient als wesentlicher Schutz gegen Verunreinigungen, die die Sequenzierungsergebnisse verfälschen könnten.
4. Was sollte bei der Datenanalyse für Ultra Low RNA-Sequenzierung berücksichtigt werden?
Bei der Datenanalyse in der Ultra Low RNA-Sequenzierung wird eine sorgfältige Datenvorverarbeitung unerlässlich, um die Qualität zu validieren und das Rauschen zu minimieren, was die Präzision in nachfolgenden Analysen erhöht. Die Bewertung der Genexpressionsniveaus konzentriert sich auf eine genaue Quantifizierung, während eine tiefgehende Analyse der funktionalen Annotation dazu beiträgt, die biologische Bedeutung von differentiell exprimierten Genen zu entschlüsseln. Die Integrationsanalyse erweitert die Erkenntnisse zusätzlich, indem sie kombiniert. RNA-Sequenzierung Daten mit anderen Omics-Datensätzen zu kombinieren, um ein umfassendes Verständnis biologischer Mechanismen zu ermöglichen.
SMART-seq Fallstudien
Bewertung von RNA-Sequenzierung mit extrem niedriger Eingabe zur Untersuchung des menschlichen T-Zell-Transkriptoms
Journal: Wissenschaftliche Berichte
Impactfaktor: 4,996
Veröffentlicht: 11. Juni 2019
Zusammenfassung
Das Verständnis der T-Zell-Biologie ist entscheidend für die therapeutische Entwicklung. Diese Studie bewertete Methoden der RNA-Sequenzierung mit ultra-niedrigem Input und stellte fest, dass AmpliSeq konsistent kodierende Gene selbst bei niedrigen Inputs nachwies, während SMART nicht-kodierende Gene detektierte, jedoch bei sehr niedrigen Inputs Variabilität zeigte. Alle Methoden identifizierten Aktivierungssignaturen von T-Zellen bei 1.000 Zellen, wobei AmpliSeq bei 100 Zellen besser abschnitt.
Materialien & Methoden
- Blutprobe des Menschen
- T-Zell-Isolierung
- RNA-Extraktion
- cDNA-Synthese
- Niedriginput-Bibliotheksvorbereitung
- Whole Transcriptom-Sequenzierung
- Gezielte Transkriptom-Sequenzierung
- Illumina HiSeq2000/NextSeq500
- Ausrichtung an das menschliche Genom
- Differenzielle Genexpressionsanalyse
- Weganalyse
- Heatmap-Visualisierung
Ergebnisse
Die Autoren evaluierten drei RNA-Extraktionskits (PicoPure, Zymogen, Qiagen RNeasy Mikro-Kit) an primären menschlichen naiven CD4 T-Zellen (5K, 1K, 100 Zellen). Das Qiagen RNeasy Mikro-Kit bot die beste Konsistenz, insbesondere bei 100 Zellen, und wurde für weitere Studien ausgewählt. T-Zellen, die mit α-CD3 oder α-CD3 und B7-1 Fc behandelt wurden, wurden zur RNA-Bibliotheksvorbereitung mit SMART-Seq (Nextera: SMART_Nxt, Clontech: SMART_CC) und AmpliSeq-Technologien verwendet. AmpliSeq zeigte höhere Alignierungsraten (81-92%) als SMART-Seq (59-74%), mit stabiler Gen-Detektion über verschiedene Eingaben hinweg. SMART-Seq detektierte weniger Gene bei abnehmenden Eingaben und konnte nicht-kodierende Gene nachweisen, was AmpliSeq nicht bewerten kann.
Die Analyse der differentiellen Genexpression in RNA-seq liefert entscheidende Einblicke in biologische Prozesse. Unter Verwendung von 100 K Zellproben als Referenz identifizierten SMART_Nxt, SMART_CC und AmpliSeq jeweils 7762, 7923 und 6662 differentielle exprimierte Gene (DEGs) zwischen den Behandlungen mit α-CD3 + B7-1 Fc und α-CD3 (FDR < 0,05). 4261 DEGs waren plattformübergreifend gemeinsam und zeigten konsistente Faltungsänderungen (R2 = 0,94 für SMART_Nxt/SMART_CC, R2 = 0,84 für AmpliSeq/SMART_CC, R2 = 0,85 für SMART_Nxt/AmpliSeq). Plattform-spezifische DEGs wiesen unterschiedliche Expressionsmuster und Variabilität zwischen technischen Replikaten auf.
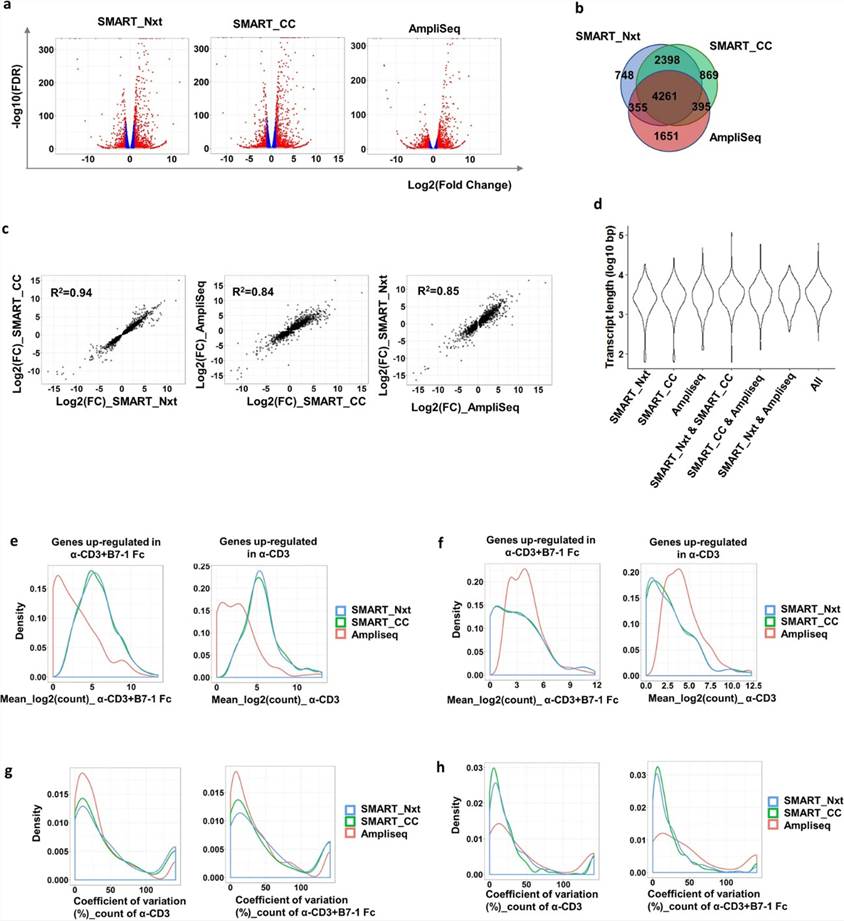 Abb. 1. Drei verschiedene Plattformen identifizierten gemeinsame unterschiedlich exprimierte Gene; jedoch wurde auch plattformspezifische Erkennung beobachtet.
Abb. 1. Drei verschiedene Plattformen identifizierten gemeinsame unterschiedlich exprimierte Gene; jedoch wurde auch plattformspezifische Erkennung beobachtet.
Die Autoren bewerteten die DEG-Erkennung bei unterschiedlichen Zellinputs und stellten fest, dass AmpliSeq konsequent mehr DEGs als SMART_Nxt und SMART_CC bei niedrigeren Zellinputs identifizierte, wobei die Anzahl der DEGs mit abnehmenden Inputmengen korrelierte. Zum Beispiel detektierte AmpliSeq 6662 DEGs bei 100 K Zellen, 3382 bei 5 K, 844 bei 1 K und 90 bei 100 Zellen, was einen signifikanten Rückgang der DEG-Zahlen bei abnehmendem Zellinput zeigt. AmpliSeq zeigte auch eine höhere Sensitivität bei der Erkennung signifikanter DEGs bei niedrigeren Inputs im Vergleich zu SMART_Nxt und SMART_CC. Darüber hinaus offenbarte GSEA eine Anreicherung von Signalwegen, die mit der T-Zell-Aktivierung in Proben behandelt mit α-CD3 + B7-1 Fc in Verbindung stehen, und hob biologische Prozesse hervor, die von der Zellaktivierung betroffen sind.
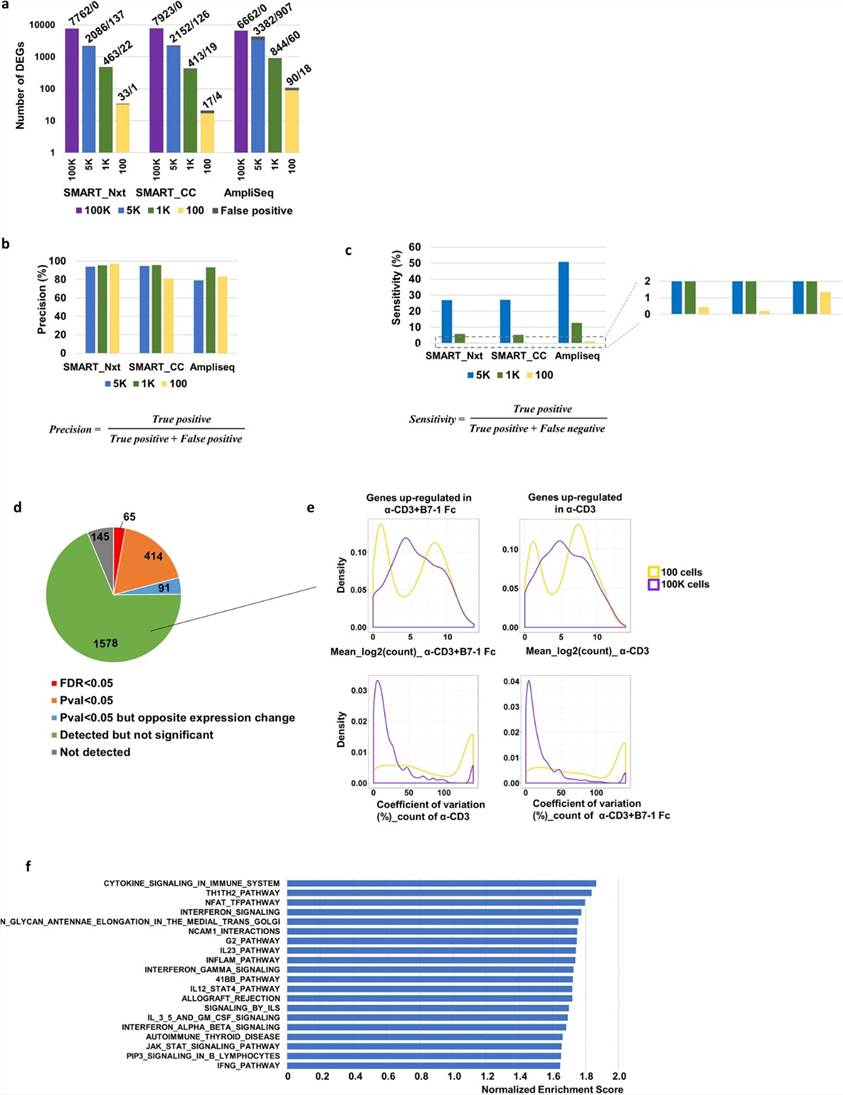 Abb. 2. Die Anzahl der differentially expressed genes nahm mit reduziertem Input ab, die Präzision der Detektion blieb robust und AmpliSeq zeigte eine höhere Sensitivität.
Abb. 2. Die Anzahl der differentially expressed genes nahm mit reduziertem Input ab, die Präzision der Detektion blieb robust und AmpliSeq zeigte eine höhere Sensitivität.
In dieser Studie verglichen die Autoren 15 Gene, die an der T-Zell-Aktivierung beteiligt sind, über verschiedene Technologien hinweg. Bei höheren Eingaben (100 K, 5 K, 1 K) schnitten alle Technologien ähnlich gut ab, wenn es darum ging, Veränderungen der Genexpression zu erkennen. Bei einer Eingabe von 100 Zellen identifizierte AmpliSeq eine signifikante Hochregulation von 7 Genen (CD200, CD69, EGR1, FASLG, IL2, MYC, TNF), während SMART_Nxt und SMART_CC weniger Gene erkannten (jeweils 1). Bemerkenswert ist, dass TNFSF14 nur von SMART_Nxt und SMART_CC erkannt wurde, selbst bei höheren Eingaben. qRT-PCR bestätigte die differentielle Expression von 12 von 16 Genen und hob Probleme bei der IL-21-Detektion hervor, die möglicherweise auf Primerbeschränkungen zurückzuführen sind.
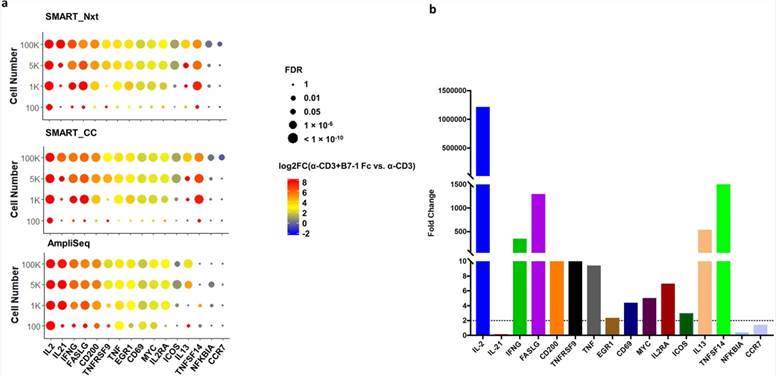 Abb. 3. Die Erkennung von bekannten T-Zell-Aktivierungsmarkern wurde bei einem Zelleneingang von 1 K und darüber erreicht, und AmpliSeq detektierte einen höheren Prozentsatz dieser Gene bei einem Zelleneingang von 100 Zellen.
Abb. 3. Die Erkennung von bekannten T-Zell-Aktivierungsmarkern wurde bei einem Zelleneingang von 1 K und darüber erreicht, und AmpliSeq detektierte einen höheren Prozentsatz dieser Gene bei einem Zelleneingang von 100 Zellen.
Fazit
In dieser Studie wurden RNA-seq-Technologien (SMART_Nxt, SMART_CC und AmpliSeq) zur Untersuchung von Veränderungen der Genexpression in T-Zellen während der Aktivierung evaluiert. AmpliSeq zeigte eine überlegene Leistung beim Nachweis von Genen über verschiedene Eingangslevel hinweg und behielt die Sensitivität für niedrig exprimierte Gene bei. Allerdings wiesen SMART_Nxt und SMART_CC eine höhere Übereinstimmung miteinander beim Nachweis spezifischer Gene auf, möglicherweise aufgrund ihrer gemeinsamen SMART-Technologie. Bei extrem niedrigen Eingaben (100 Zellen) zeigte AmpliSeq eine bessere Sensitivität beim Nachweis biologisch relevanter Veränderungen der Genexpression im Vergleich zu den SMART-Technologien.
Referenz:
- Wang, J., Rieder, S.A., Wu, J. et al. Bewertung der RNA-Sequenzierung mit ultra-niedrigem Input zur Untersuchung des menschlichen T-Zell-Transkriptoms. Wissenschaftliche Berichte, 2019, 9, 8445.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Chaperon-vermittelte Autophagie steuert proteomische und transkriptomische Wege zur Aufrechterhaltung der Aktivität von Gliom-Stammzellen.
Zeitschrift: Krebsforschung
Jahr: 2022
Zirkuläre DNA-Tumorviren erzeugen zirkuläre RNAs.
Zeitschrift: Mitteilungen der Nationalen Akademie der Wissenschaften
Jahr: 2018
Wiederholte Immunisierung mit ATRA-haltigem liposomalem Adjuvans transdifferenziert Th17-Zellen zu einem Tr1-ähnlichen Phänotyp.
Zeitschrift: Zeitschrift für Autoimmunität
Jahr: 2024
Die Rolle der Histonvariante H2A.Z.1 in Gedächtnis, Transkription und alternativer Spleißung wird durch Lysinmodifikationen vermittelt.
Zeitschrift: Neuropsychopharmakologie
Jahr: 2024
FAK-Verlust reduziert die ERK-Phosphorylierung, die durch BRAFV600E induziert wird, um die intestinale Stammzelligkeit und die Bildung von Blinddarmtumoren zu fördern.
Journal: Elife
Jahr: 2023
Identifizierung von zirkulären RNAs, die die Proliferation von Kardiomyozyten in neonatale Schweineherzen regulieren
Journal: JCI Insight
Jahr: 2024
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


