
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Virale Metagenomische Sequenzierung
Als erfahrener Anbieter von NGS-Diensten und Partner von Illumina verpflichtet sich CD Genomics, beispiellose Mengen an Sequenzdaten anzubieten, die schnelle und innovative Analyseansätze mit kosteneffizienten Lösungen ermöglichen. Wir verfügen über umfangreiche Fachkenntnisse in der Bereitstellung eines zuverlässigen und unvoreingenommenen viralen metagenomischen Sequenzierungsdienstes, indem wir modernste Hochdurchsatz-Sequenzer, Sequenzierungsstrategien und Bioinformatik Pipelines.
Einführung in die virale Metagenom-Sequenzierung
Viren sind die häufigsten biologischen Entitäten der Welt, die in den meisten Umgebungen mikrobiellen Zellen im Verhältnis 10:1 übertreffen. Viren bewohnen ständig unseren Körper, und asymptomatische Wirte sind keine Ausnahme. Diese aufkommende Sichtweise erfordert die Erforschung des Viroms. Virale Metagenomik kann Einblicke in die Zusammensetzung und Struktur viraler Gemeinschaften bieten. Die Profilierung der taxonomischen Zusammensetzung viraler Gemeinschaften ist nicht nur für die Grundlagenforschung, sondern auch für die klinische Wissenschaft und Praxis wichtig.
Viralgemeinschaften sind jedoch schwer zu charakterisieren, da es kein einzelnes Gen gibt, das von allen viralen Genomen geteilt wird, was die Anwendung analoger Methoden, die bei Bakterien für das ribosomale DNA-Profiling verwendet werden, einschränkt. Die geringe Menge an freier und zellulärer DNA ist eine weitere Herausforderung. Metagenomisches Shotgun-Sequencing und RNA-Seq Es gibt zwei Ansätze zur Charakterisierung von viralen Gemeinschaften. Die direkte Sequenzierung kann zu einem hohen Hintergrund an genetischem Material führen. Daher sind zusätzliche Verfahren erforderlich, um virale Partikel (VPs) zu konzentrieren und zu reinigen. Es wurden mehrere praktikable Anreicherungsmethoden vorgeschlagen, darunter Filtration, Ultrazentrifugation, Nucleasenbehandlung, multiple Displacement-Amplifikation (MDA), linker-amplifizierte Shotgun-Bibliothek (LASL) und zufälliger PCR-Ansatz.
Vorteile der viralen Metagenom-Sequenzierung
- Keine Notwendigkeit für Kultivierung oder Antikörperlaboruntersuchungen.
- Umfassende Informationen über die Biodiversität der Gemeinschaft, Taxonomie und Funktion
- Vielversprechende Anwendungen umfassen die Entdeckung von Viren und die Identifizierung viraler Krankheitserreger, die mit Gartenbau, Tiermedizin und menschlicher Gesundheit verbunden sind, insbesondere bei schwer zu diagnostizierenden Fällen.
Anwendung der viralen Metagenom-Sequenzierung
(1) Krankheitsbehandlung: Das Studium der viralen Vielfalt, die Entdeckung unbekannter Viren, die Detektionsanalyse spezifischer Viren/Bakteriophagen und die Interaktion zwischen Bakterien und Bakteriophagen (in Aspekten wie dem Darm, der Haut, der Umwelt usw. und in Bezug auf die Gesundheit) sind entscheidende Elemente, die angegangen werden müssen. Die Untersuchung der Beziehungen zwischen Viren und Krankheiten (wie Stoffwechselerkrankungen, Magen-Darm-Erkrankungen, Atemwegserkrankungen, Allergien, Infektionskrankheiten, Mundkrankheiten und Tumoren) kann unser Verständnis erheblich erweitern. Techniken wie die Bakteriophagentherapie und die Sensibilisierung von antibiotikaresistenten Bakterien könnten potenziell Immunantworten stimulieren.
(2) Lebensmittelsicherheit: Die Überwachung von Viren in Lebensmitteln, das Verständnis der viralen Diversität in Lebensmitteln, die Untersuchung von über Lebensmittel übertragenen Krankheitserregern und die Überwachung von Viren während der Lebensmittelverarbeitung sind entscheidende Maßnahmen. Die Rückverfolgung von Vorfällen mit viraler Kontamination und das Studium des Zusammenhangs zwischen Viren und dem Lebensmittelmikrobiom können erheblich zur Gewährleistung der Lebensmittelsicherheit beitragen.
(3) Viehzuchtindustrie: Das Management von Krankheiten in der Tierzucht, wie der Umgang mit Durchfall, Schweinepest, Maul- und Klauenseuche, zoonotischen Krankheiten und die Analyse von Fäkalien von Wildtieren, ist entscheidend für die Erhaltung der Gesundheit von Nutztieren.
(4) Überwachung von Infektionskrankheiten: Die Überwachung von Umweltviren kann frühzeitige Erkennung und Hinweise auf Krankheitsausbrüche liefern und somit erheblich zu öffentlichen Gesundheitsinitiativen beitragen.
(5) Umwelttechnik: In der Umwelttechnik, wie beispielsweise bei der Abwasserbehandlung und der Schadstoffkontrolle, hilft das Verständnis von viralen Gemeinschaften in Gewässern, die Wirksamkeit von Behandlungen und die Umweltauswirkungen zu bewerten. Dies gilt für verschiedene Umgebungen wie öffentliche Verkehrssysteme, Krankenhäuser, Ozeane, Süßwassergewässer, Luft und Gletscher.
(6) Arzneimittelentdeckung: Die Erforschung des Viroms kann Viren aufdecken, die mit Wirten interagieren könnten, und Hinweise für die Entdeckung neuer Medikamente liefern.
(7) Landwirtschaft: Das Studium der viralen Vielfalt von Pflanzen und Böden könnte die Entwicklung von Biopestiziden im Agrarsektor unterstützen.
Viral Metagenomische Sequenzierungs-Workflow
Unser hochqualifiziertes Expertenteam führt das Qualitätsmanagement durch, indem es jedes Verfahren befolgt, um umfassende und genaue Ergebnisse sicherzustellen. Der allgemeine Arbeitsablauf für virale metagenomische Shotgun-Sequenzierung ist unten skizziert. Kurz gesagt, die grundlegenden Schritte, die an der viralen Metagenomik beteiligt sind, umfassen die Isolation und Reinigung von viralen Partikeln durch Größenfiltration oder dichtebasierte Zentrifugation, sequenzunabhängige Amplifikation von viraler Nukleinsäure, Shotgun-Sequenzierung oder RNA-Seq und abgestimmte Analysen zu Vielfalt, Taxonomie und Funktion durch eine Reihe zuverlässiger bioinformatischer Werkzeuge.

Dienstspezifikationen
Musteranforderungen
|
|
 |
Sequenzierungsplattformen
|
 |
BioinformatikanalyseWir bieten mehrere maßgeschneiderte bioinformatische Analysen an:
|
Analyse-Pipeline
Liefergegenstände
- Qualitätskontrolle und Hostentfernung
- Analyse der Lesartenarten
- Versammlung
- Analyse der zusammengesetzten Arten
- Funktionalanalysis
- Vorhersage von Phagenwirten
Neben der qualifizierten viralen metagenomischen Sequenzierung bieten wir Unterstützung an, einschließlich des experimentellen Designs, der Bestimmung der geeigneten Sequenzierungsplattform, Software-Tools und Analysemethoden, um Ihr Projekt zu unterstützen. Wenn Sie zusätzliche Anforderungen oder Fragen haben, zögern Sie bitte nicht, uns zu kontaktieren. Unsere erfahrenen Spezialisten helfen Ihnen gerne, Ihre Fragen zu klären.
Referenzen:
- Li Y, et al. VIP: eine integrierte Pipeline für die Metagenomik zur Identifizierung und Entdeckung von Viren. Wissenschaftliche Berichte, 2016, 6: 3774
- Rampelli, S. et al. ViromeScan: ein neues Werkzeug zur metagenomischen Profilierung viraler Gemeinschaften. BMC Genomik2016, (17):165.
- Lewandowska D W, Zagordi O, Geissberger F D, et al. Optimierung und Validierung der Probenvorbereitung für die metagenomische Sequenzierung von Viren in klinischen Proben. Mikrobiom, 2017, 5(1): 94.
Demonstrationsergebnisse
Teilweise Ergebnisse sind unten aufgeführt:
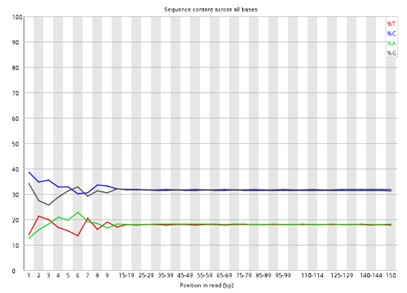
Pro Basissequenzinhalt.
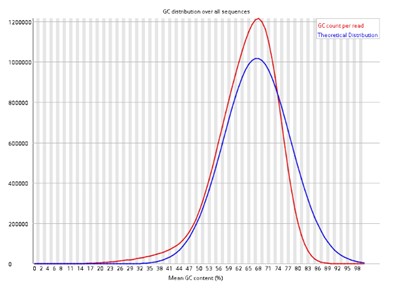
Pro Sequenz GC-Gehalt.
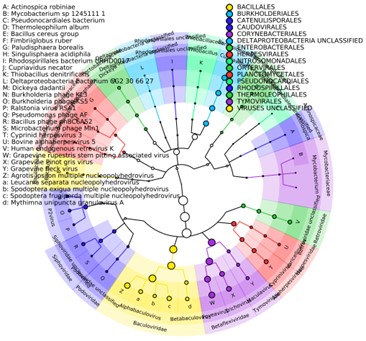
Vereinigte_Überfluss.
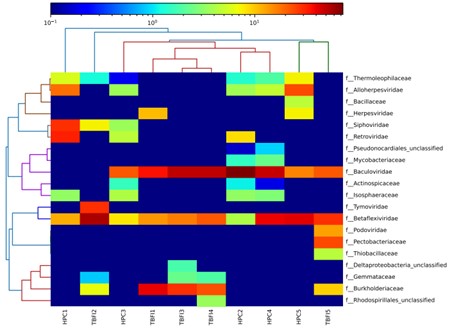
Abundanz-Hitzekarte auf Familienebene.
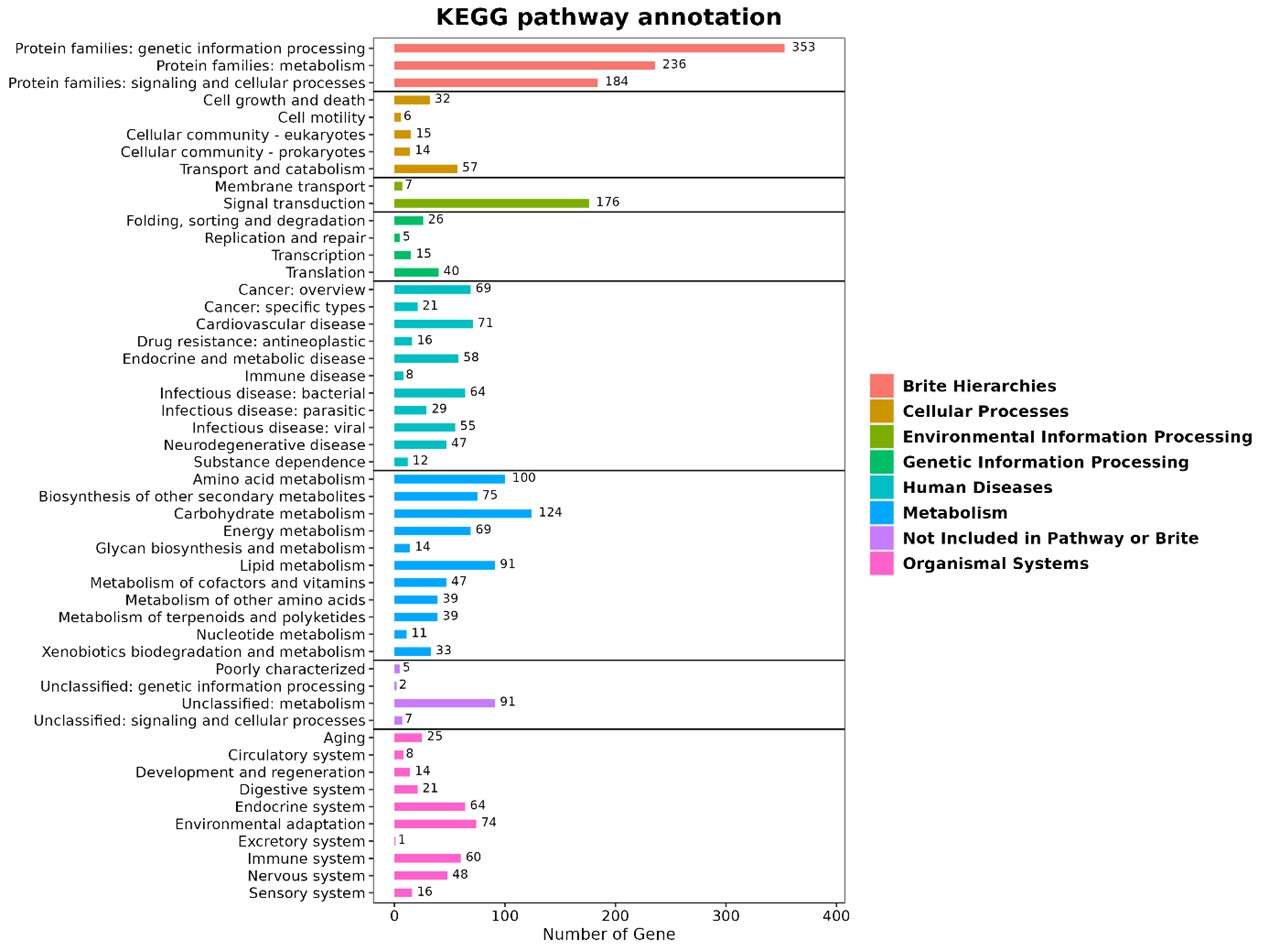
KEGG-Klassifikation.
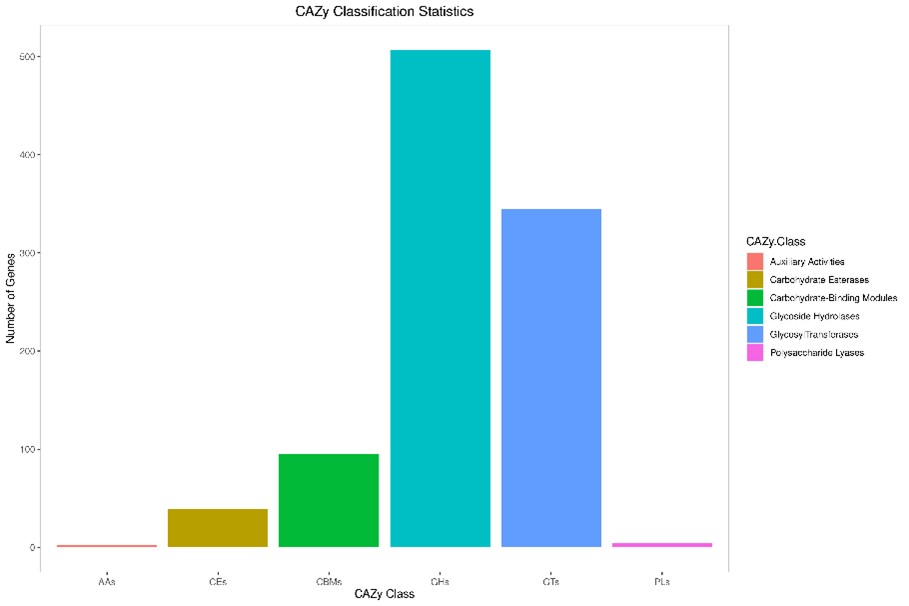
CAZy-Funktionsklassifikation.
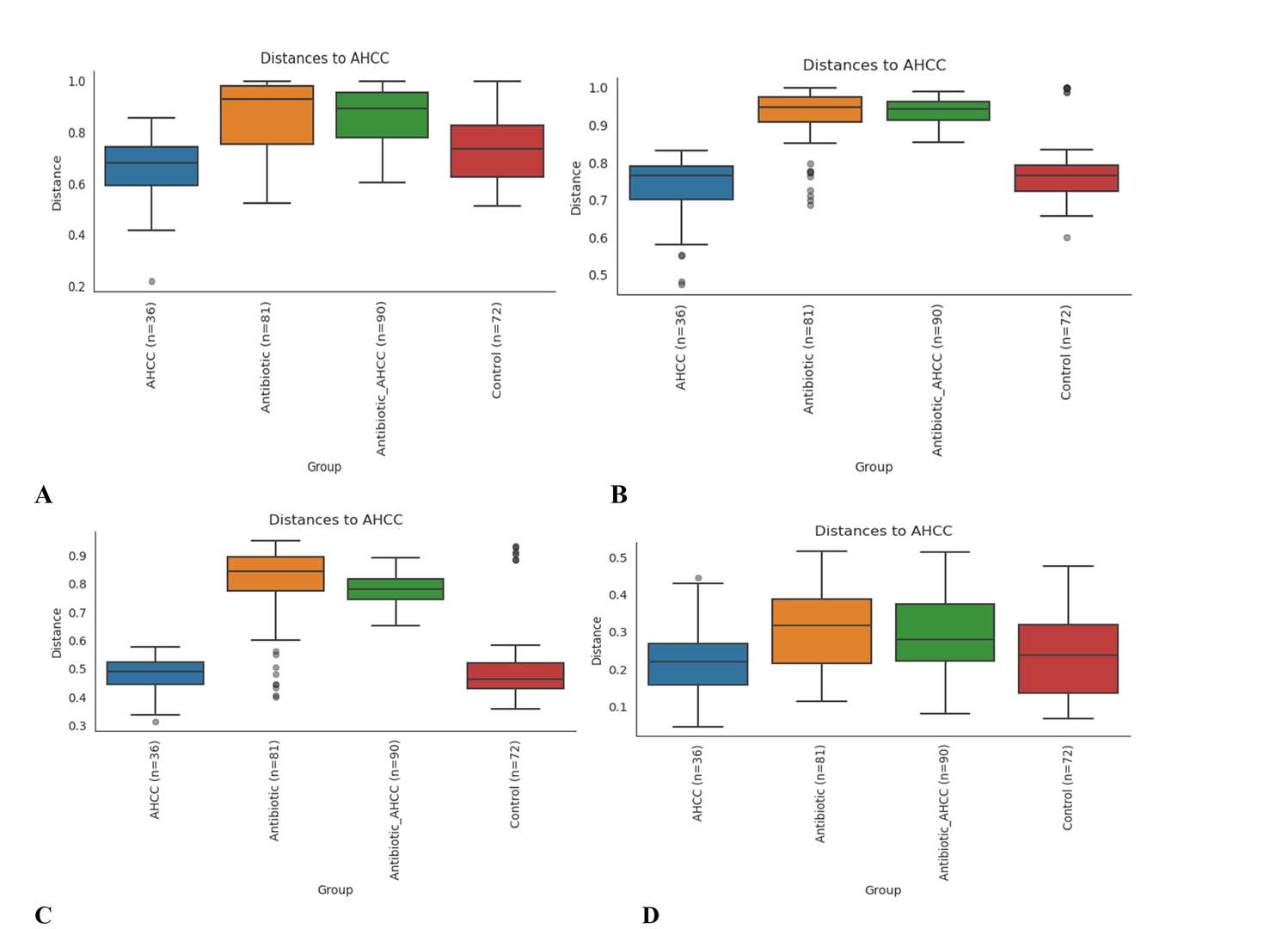
Boxplot-Analyse basierend auf Bray-Curtis (A), binärem Jaccard (B), ungewichteten Unifrac (C) und gewichteten Unifrac (D).
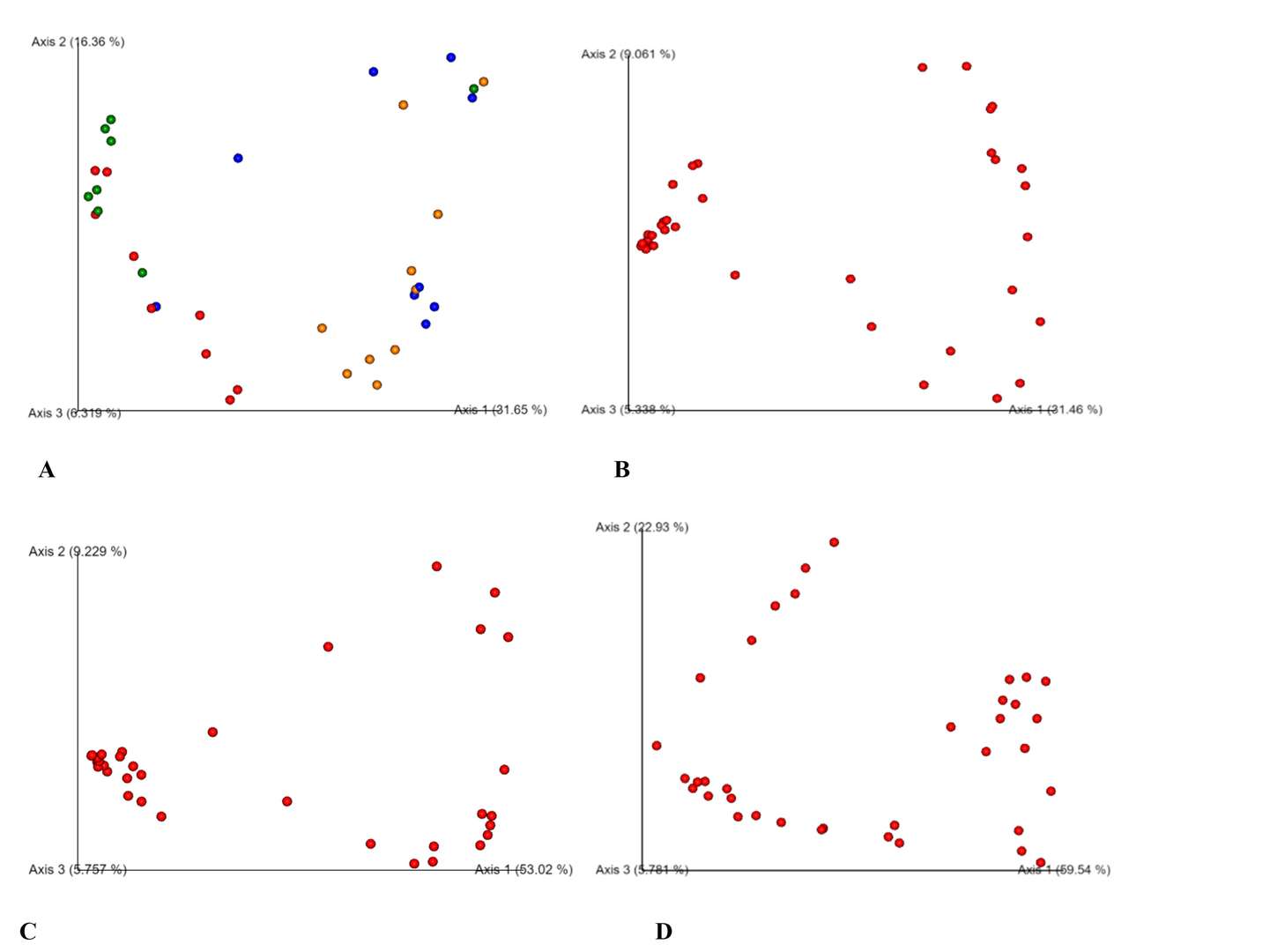
PCoA-Analyse basierend auf Bray-Curtis (A), binärem Jaccard (B), ungewichteten Unifrac (C) und gewichteten Unifrac (D).
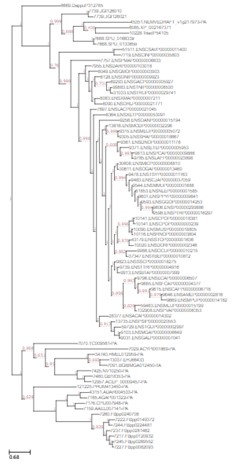
UPGMA-Hierarchiebaum.
Viral Metagenomische Sequenzierungs-FAQs
1. Welche Ansätze gibt es für die Ganzgenomsequenzierung von Krankheitserregern?
Derzeit gibt es drei Hauptansätze zu Whole-Genome-Sequenzierung von Krankheitserregern, d.h., metagenomische Sequenzierung, PCR-Amplicon-Sequenzierung und Zielanreicherungssequenzierung (Abbildung 1). Die direkte metagenomische Sequenzierung bietet eine genaue und integrierte Darstellung der Sequenzen innerhalb einer Probe. Die PCR-Amplicon-Sequenzierung führt PCR-Reaktionen durch, um das virale Genom anzureichern, was die Arbeitslast für große Genome erheblich erhöht, aber die Kosten senkt. Die Zielanreicherungssequenzierung verwendet virus-spezifische Nukleotid-Sonden, die an einer festen Phase gebunden sind, um das virales Genom In einer einzigen Reaktion, die die Arbeitsbelastung verringert, aber die Kosten im Vergleich zur PCR-Amplicon-Sequenzierung erhöht. Die Vor- und Nachteile sind in Tabelle 1 aufgeführt. Sie zeigt, dass die virale metagenomische Sequenzierung die einzigartigen Vorteile bietet, Pathogene zu untersuchen und die virale Vielfalt in Umwelt- und klinischen Proben zu charakterisieren.
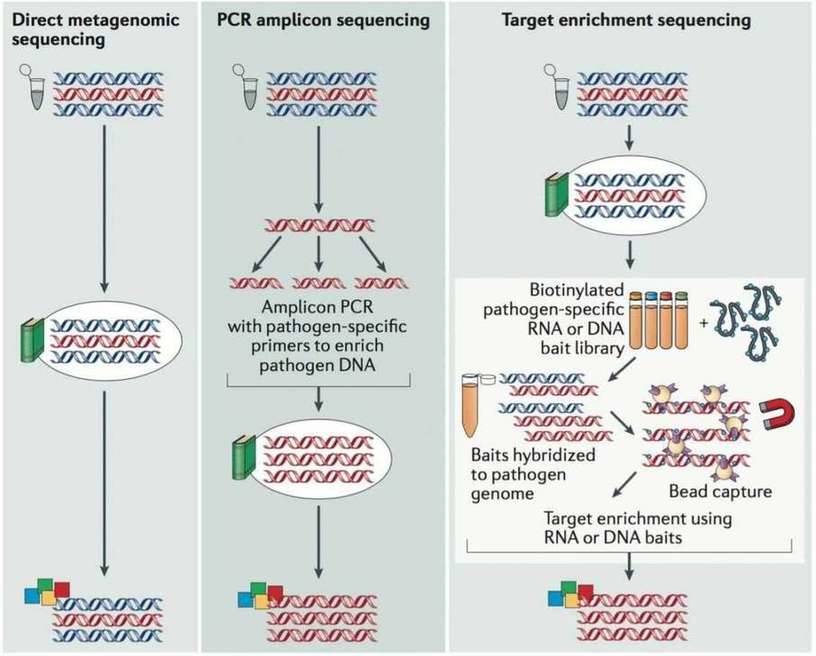 Abbildung 1. Die Hauptansätze zur Sequenzierung viraler Genome (Houldcroft et al. 2016).
Abbildung 1. Die Hauptansätze zur Sequenzierung viraler Genome (Houldcroft et al. 2016).
Tabelle 1. Vorteile und Nachteile verschiedener Methoden zur Sequenzierung viraler Genome (Houldcroft et al. 2016).
| Methode | Vorteile | Nachteile |
|---|---|---|
| Metagenomik |
|
|
| PCR-Amplikon |
|
|
| Zielanreicherung |
|
|
2. Was ist der allgemeine Bioinformatik-Workflow für die virale Metagenom-Sequenzierung?
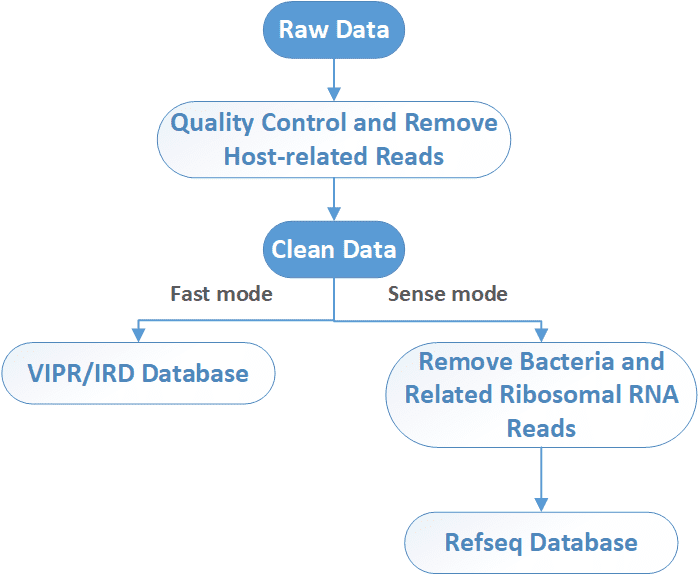
Nach der Sequenzierung werden die Roh-NGS-Reads zunächst durch die Entfernung von Adaptern, niedrigqualitativen und niedrigkomplexen Sequenzen vorverarbeitet, gefolgt von einer rechnergestützten Subtraktion der hostbezogenen Reads. In einem schnellen Modus werden Viren durch Alignment an die ViPR/IRD Nucleotid-Datenbank identifiziert. In einem sinnvolleren Modus werden Bakterien und verwandte ribosomale RNA-Reads entfernt, bevor das Alignment an die Virusdatenbank erfolgt. Nicht zugeordnete Reads werden weiter an eine virale Proteindatenbank (NCBI RefSeq DB) ausgerichtet. Anschließend erfolgt die taxonomische Identifizierung (Taxl), das Abdeckungsdiagramm (Covplot), von Neuem Die Assemblierung (mehrere k-Mer) und die phylogenetische Analyse (PhyGo) können durchgeführt werden.
Referenzen:
- Houldcroft C J, Beale M A, Breuer J. Klinische und biologische Erkenntnisse aus der viralen Genomsequenzierung. Nature Reviews Microbiology, 2017, 15(3): 183-192.
- Li Y, Wang H, Nie K, et al. VIP: eine integrierte Pipeline für die Metagenomik zur Identifizierung und Entdeckung von Viren. Scientific Reports, 2016, 6.
Viral Metagenomische Sequenzierungs-Fallstudien
Nachweis und Sequenzierung des Zika-Virus aus Fruchtwasser von Föten mit Mikrozephalie in Brasilien: eine Fallstudie
Zeitschrift: The Lancet Infectious Diseases
Impactfaktor: 19,864
Veröffentlicht: 17. Februar 2016
Hintergründe
Die Fälle von Mikrozephalie in Brasilien im Jahr 2015 waren 20 Mal höher als in den Vorjahren. Epidemiologische Daten deuten darauf hin, dass die Inzidenz von Mikrozephalie in Brasilien möglicherweise mit dem Zika-Virus in Verbindung steht. Die Autoren entnahmen Fruchtwasserproben von zwei schwangeren Frauen in Brasilien, deren Föten mit Mikrozephalie diagnostiziert wurden, um die Ursache der Mikrozephalie zu untersuchen.
Methoden
- Schwangere Frauen mit Symptomen einer Zika-Virus-Infektion
- Feten, bei denen Mikrozephalie diagnostiziert wurde
- 5 ml Amnionflüssigkeitsproben von zwei Frauen in der 28. Schwangerschaftswoche
- Serologietests
- ELISA
- Reinigung von Viruspartikeln
- Quantitative umgekehrte Transkriptions-PCR
- Virale metagenomische Sequenzierung
- PRINSEQ
- BLAST
- RefSeq
- De novo Versammlung
- Phylogenetische Analyse
Ergebnisse
1. Zika-Virus-Genomassemblierung
Nach der Entfernung zellulärer Kontamination wiesen 288.904 Sequenzen Ähnlichkeiten mit Virusgenomen auf, und 683 Sequenzen stimmten mit dem Zika-Virusgenom überein. Abbildung 1 zeigt das gesamte Zika-Virusgenom, das aus Patient 1 isoliert wurde, mit Genannotationen.
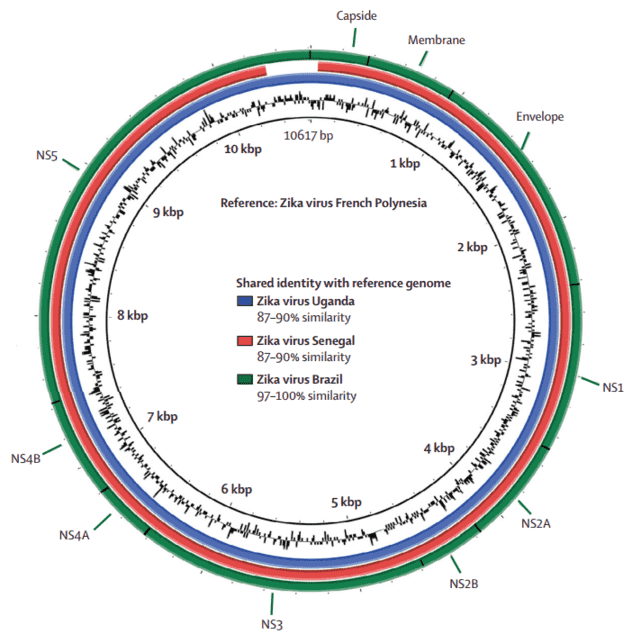 Abbildung 1. Vergleichendes Genome-BLAST-Atlas-Diagramm des Zika-Virus. Der grüne Kreis entspricht dem vollständigen brasilianischen Zika-Virus, das von Patient 1 isoliert wurde. Der rote Kreis entspricht dem senegalesischen Stamm des Zika-Virus. Der blaue Kreis entspricht dem ugandischen Stamm.
Abbildung 1. Vergleichendes Genome-BLAST-Atlas-Diagramm des Zika-Virus. Der grüne Kreis entspricht dem vollständigen brasilianischen Zika-Virus, das von Patient 1 isoliert wurde. Der rote Kreis entspricht dem senegalesischen Stamm des Zika-Virus. Der blaue Kreis entspricht dem ugandischen Stamm.
2. Phylogenetische Analyse
Die phylogenetischen Analysen wurden mit dem kodierenden Bereich für die Hüllgene (Abbildung 2) und NS5-Gene (Abbildung 3) durchgeführt. Die geographische Region des brasilianischen Zika-Virus-Stammes konnte aufgrund von Probenbeschränkungen nicht bestimmt werden, schien jedoch enger mit dem französisch-polynesischen Stamm als mit afrikanischen Stämmen verwandt zu sein.
Die geografischen und chronologischen Verteilungen der Zika-Virus-Linien deuteten darauf hin, dass der südostasiatische Stamm sich genetisch vor etwa 50 Jahren von den afrikanischen Stämmen isoliert haben könnte. Dieses Muster wurde durch die genetische Distanz zwischen den neuen brasilianischen Zika-Virus-Sequenzen und dem ugandischen Zika-Virus-Genom weiter bestätigt (Abbildung 4).
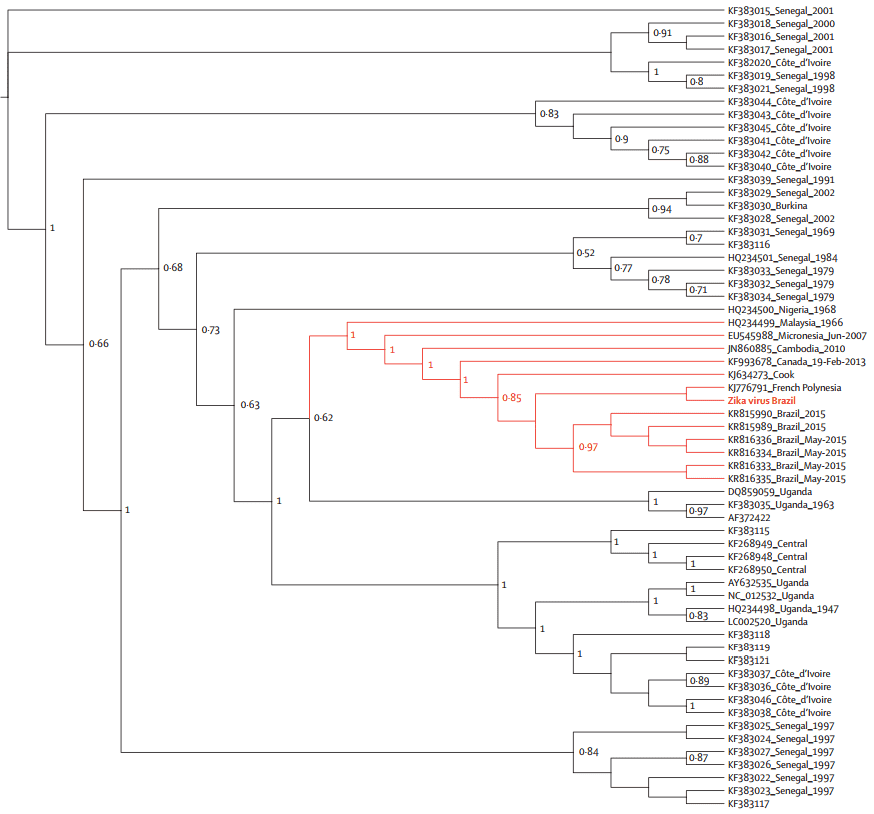 Abbildung 2. Maximum-Likelihood-Topologien des Hüllengenombereichs des brasilianischen Zika-Virus.
Abbildung 2. Maximum-Likelihood-Topologien des Hüllengenombereichs des brasilianischen Zika-Virus.
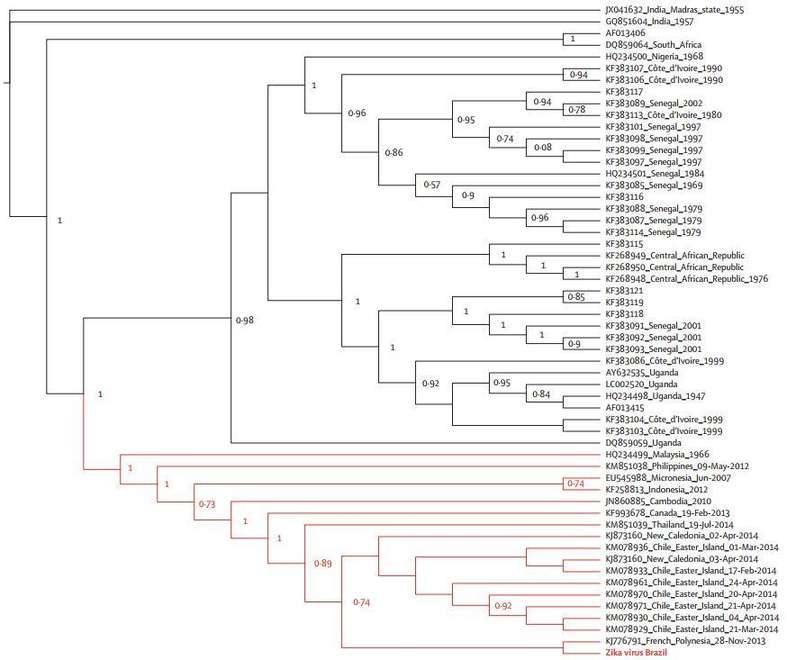 Abbildung 3. Maximum-Likelihood-Topologien des NS5-Genombereichs des brasilianischen Zika-Virus.
Abbildung 3. Maximum-Likelihood-Topologien des NS5-Genombereichs des brasilianischen Zika-Virus.
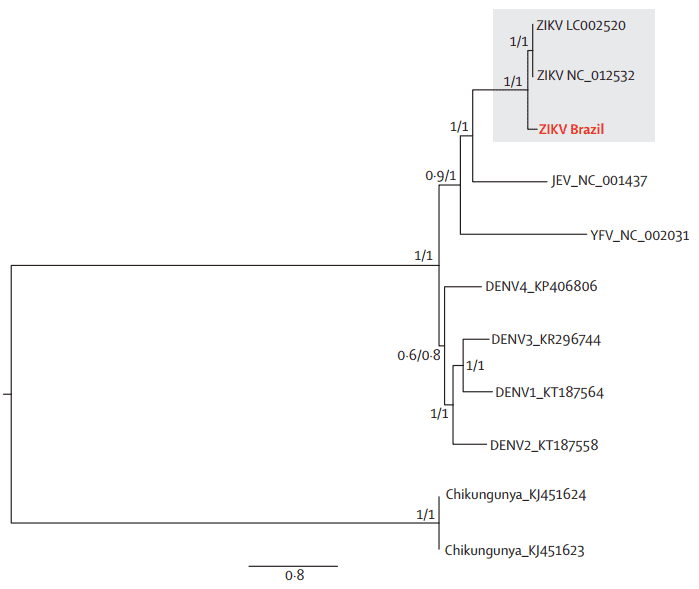 Abbildung 4. Maximum-Likelihood-Phylogenie des brasilianischen Zika-Virus, anderer Flaviviridae-Genome und eines Alphavirus-Genoms. DENV = Dengue-Virus, JEV = Japanisches Enzephalitis-Virus. YFV = Gelbfieber-Virus. ZIKA = Zika-Virus.
Abbildung 4. Maximum-Likelihood-Phylogenie des brasilianischen Zika-Virus, anderer Flaviviridae-Genome und eines Alphavirus-Genoms. DENV = Dengue-Virus, JEV = Japanisches Enzephalitis-Virus. YFV = Gelbfieber-Virus. ZIKA = Zika-Virus.
3. Eine Zika-Virus-Infektion könnte durch die transplacentare Übertragung auftreten.
Dieser Artikel war der erste, der das gesamte Genom des Zika-Virus aus Fruchtwasser isolierte, was darauf hindeutet, dass eine Zika-Virus-Infektion durch transplacentare Übertragung erfolgen könnte.
Referenz:
- Calvet G, Aguiar R S, Melo A S O, et al. Nachweis und Sequenzierung des Zika-Virus aus dem Fruchtwasser von Föten mit Mikrozephalie in Brasilien: eine Fallstudie. The Lancet Infektionskrankheiten, 2016, 16(6): 653-660.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Übertragbarer Schutz durch Darmmikroben gegen STING-assoziierte Lungenerkrankungen
Zeitschrift: Cell Reports
Jahr: 2021
Mikrobielle Anpassung und Reaktion auf hohe Ammoniakkonzentrationen und Niederschläge während der anaeroben Vergärung unter psychrophilen und mesophilen Bedingungen
Zeitschrift: Wasserforschung
Jahr: 2021
Algen-bakterielle Synergie bei der Behandlung von Weinkellerei-Abwasser
Journal: NPJ Sauberes Wasser
Jahr: 2018
Biokonversion von Schwarzen Soldatenfliegen zu Medienkomponenten für kultiviertes Fleisch unter Verwendung des Mikrobioms des Darms von Blaukanälen.
Journal: Bioresource Technology Berichte
Jahr: 2024
Indol-3-Propionsäure, ein Metabolit der Darmmikrobiota, schützt vor der Entwicklung von postoperativem Delirium.
Zeitschrift: Annalen der Chirurgie
Jahr: 2023
Erläuterung der Auswirkungen von biologischen vs. konventionellen Anbaumethoden und Rhizobien-Inokulation auf die mikrobielle Vielfalt im Rhizosphäre und den Ertrag von Erdnüssen.
Journal: Umweltmikrobiom
Jahr: 2023
Mehr ansehen Artikel, die von unseren Kunden veröffentlicht wurden.


